美国食品药品监督管理局FDA新指南给全球癌症药物研发带来巨大影响

重磅!美国食品药品监督管理局(FDA)建议企业在临床试验中,在可行情况下将总生存期(OS)作为主要终点。这一新指南的出台,正值CBER主任文莱·普拉萨德(Vinay Prasad)意外回归之际,他此前曾主张优先考虑总生存期(OS)。
FDA最新指南草案强调在癌症药物临床试验中,在可行的情况下应优先采用总生存期(OS) 作为主要终点,这可能会增加企业获得加速批准的难度。这一政策变化尤其对PD-1/VEGF双抗这类热门肿瘤免疫治疗药物的研发和上市策略产生显著影响。
认识OS和替代终点
在肿瘤药物临床试验中,总生存期(Overall Survival, OS) 指的是从随机分组开始到因任何原因死亡的时间。它直接反映了治疗手段是否能真正让患者活得更长,被认为是评估抗癌药物临床获益的“金标准”和最终判断依据。
由于OS随访时间较长,且可能受到后续治疗的影响,研究人员也常使用一些能更早观察到、且与生存获益相关的替代终点(Surrogate Endpoints),例如:
无进展生存期(PFS):从随机分组开始到肿瘤发生(任何方面)进展或死亡的时间。
客观缓解率(ORR):肿瘤体积缩小达到预先规定水平的患者比例。
FDA的加速批准途径允许基于这些替代终点提前批准药物,但前提是上市后需通过进一步的确证性试验来验证其OS获益。如果确证性试验未能证实OS获益或临床获益,药物可能被撤回。
FDA新指南的核心变化
此次FDA指南草案的主要变化和强调重点在于:
优先考虑OS:明确要求在可行的情况下,应优先将OS作为随机临床试验的主要终点。这是因为OS不仅能体现疗效,也包含了治疗毒性的影响,是综合性的疗效和安全性终点。
OS数据收集成为标配:即使OS不是主要终点(例如以PFS或ORR为主要终点),FDA也强烈建议在所有随机临床试验中系统性地收集、评估和提交OS数据。在某些情况下,OS应作为预先设定的假设检验计划中的次要终点。
限制交叉(Crossover)设计的影响:指南建议限制使用交叉试验设计,以避免混淆OS分析,仅在缺乏或仅有极其有限治疗选择的疾病中允许此类设计。
加速批准路径依然存在但更审慎:FDA明确,当OS数据不明确但其他中间终点(如ORR)显示临床获益时,加速批准仍是可行的监管路径;待获得充分可靠的OS结果后,可再考虑传统批准。但隐含之意是,对加速批准后验证OS获益的要求会更严格。
新指南对PD-1/VEGF药物的影响
PD-1/VEGF靶向药物(包括双抗和联合疗法)是当前肿瘤免疫治疗的热点,主要开发领域包括非小细胞肺癌(NSCLC)、肝细胞癌、肾细胞癌等。FDA的新指南对这类药物的影响主要体现在:
研发策略前移:药企需要在更早的临床试验阶段(如II期)就开始规划和设计OS终点的评估方法,包括更长的随访时间、更精细的统计计划(如考虑非比例风险)、以及最小化缺失数据。
加速批准路径的不确定性增加:以往基于ORR或PFS获得加速批准的可能性可能会因FDA对OS的强调而降低,或者加速批准后确证OS获益的压力更大。
增加研发成本和时间:获得成熟的OS数据通常需要更长的随访时间和更大的样本量,这可能会增加临床试验的总体成本和时间投入。
差异化优势的关键:对于真正能够显著改善OS的药物,新指南有助于其在竞争中脱颖而出,形成更明确的差异化优势。
进度靠前的PD-1/VEGF(或PD-L1/VEGF)相关药物对比分析
下面表格对比了部分进度相对靠前的PD-1/VEGF(或PD-L1/VEGF)靶向药物(包括双特异性抗体和联合疗法)。需要注意的是,OS数据在许多仍在进行的临床试验中尚不成熟或未公布。
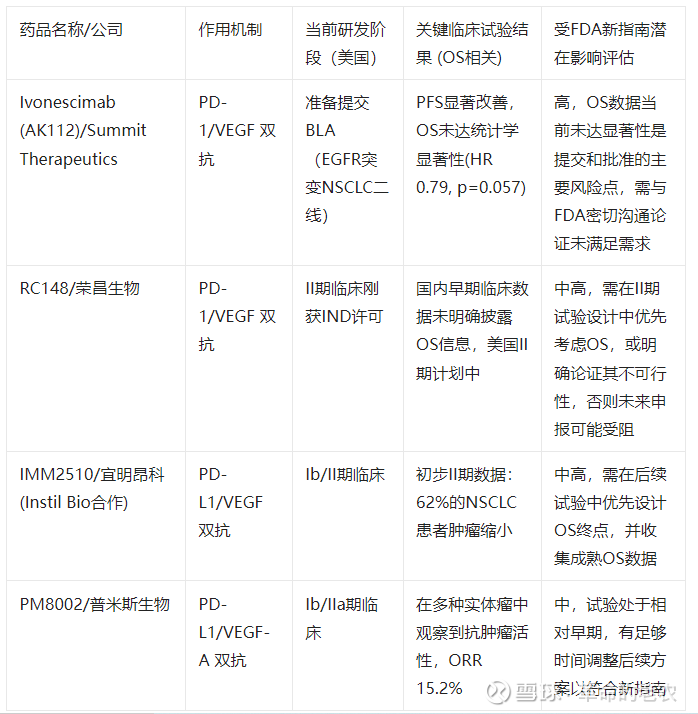
*表:部分PD-1/VEGF在研药物受FDA新指南影响分析(注:此表仅基于当前公开信息,且OS数据在许多仍在进行的试验中尚不成熟或未公布)*
说明与分析:
OS数据的成熟度是关键:对于像Ivonescimab (AK112) 这样已准备提交上市申请的产品,尽管其PFS数据优异,但OS未达到统计学显著性是一个潜在的风险点。FDA在新指南下可能会要求更长时间的随访或更多的支持性证据。
早期阶段药物的调整空间:对于多数处于I期或II期的药物(如IMM25104, PM80022, HB00257),它们有相对充足的时间来调整后续临床试验的设计,例如将OS设置为主要终点或关键次要终点,并进行更长时间的随访以收集成熟的OS数据。这可能会增加研发时间和成本,但也使得未来申报的数据更扎实。
联合疗法的挑战:对于联合治疗方案(如Pembrolizumab + Ramucirumab, Entinostat三联方案),证明其相对于标准治疗(尤其是已证明有OS获益的方案)的OS优势至关重要,且需要更大的样本量和更复杂的试验设计。
“未满足需求”领域的灵活性:在缺乏有效治疗手段的罕见肿瘤或后线治疗 setting 中,FDA基于替代终点给予加速批准的可能性仍然存在,但后续验证OS获益的要求会更严格。
生物科技企业的应对策略
面对FDA的政策变化,开发PD-1/VEGF类药物的企业可能需要:
重新评估临床试验设计:与FDA密切沟通,前瞻性地将OS纳入主要或关键次要终点并进行预设检验。
加强数据收集与管理:确保长期、高质量的OS随访数据,减少数据缺失。
探索预测性生物标志物:识别可能从治疗中获益的优势人群,富集人群,从而提高试验效率并获得更显著的疗效数据(包括OS)。
权衡加速批准与完全批准路径:评估基于替代终点寻求加速批准的风险与收益,确保确证性试验能够顺利开展并达到OS终点。
给投资者的建议
FDA的监管风向强调OS数据,这增加了肿瘤药物研发的难度和不确定性,但长远看有助于提升上市药物的质量。
对于投资者而言,关注在研PD-1/VEGF药物的企业时,可以重点考察:
临床试验终点设计:是否前瞻性地将OS纳入主要或关键次要终点并进行预设检验。
与监管机构的沟通:是否与FDA等监管机构保持密切沟通,就试验设计达成一致。
OS数据的成熟度与趋势:即使目前未达到统计学显著性,积极的OS趋势也很重要。
财务实力与研发耐力:能否支持需要更长时间和更大规模的确证性临床试验。
综上所述:
FDA拟议的新指南标志着肿瘤药物开发更加注重确凿的、以患者为中心的临床获益证据。对于PD-1/VEGF这类热门靶点药物而言,意味着“加速批准”的门槛提高,未来OS数据将成为“硬通货”。
这要求药企重新审视研发策略,更大程度地将OS获益融入药物开发计划。对于那些能够真正改善患者生存、并能量化证明这一点的药物来说,这一变化是一个长期利好。
我是热谈哥和革命的老农
欢迎大家关注和点评