CTLA-4与PD-1双阳Treg细胞

双重表达特征:肿瘤微环境(TME)中的Treg细胞展现出显著的免疫抑制表型,核心特征为CTLA-4的持续性高表达及PD-1的诱导性表达。数据显示,Treg细胞表面的CTLA-4表达水平约为效应T细胞的3-5倍,且不仅存在于胞膜,还大量分布于胞内储存池。尽管关于Treg是否表达PD-1曾存争议,但最新研究证实,在乳酸堆积、低氧等代谢压力下的TME中,Treg细胞会诱导表达PD-1,形成CTLA-4⁺PD-1⁺双阳性亚群。这一双阳性亚群具有更强的免疫抑制功能,通过竞争性消耗IL-2、反式内吞CD80/86以及分泌抑制性因子,构建了极具韧性的免疫逃逸屏障。
临床预后关联:CTLA-4⁺PD-1⁺双阳性Treg细胞并非偶发,而是广泛分布于非小细胞肺癌、结直肠癌、肾细胞癌及黑色素瘤等多种实体瘤的肿瘤浸润淋巴细胞(TILs)中。该亚群的丰度与疾病进展及不良预后呈显著正相关,例如在骨肉瘤中,双阳性表达直接关联肿瘤的复发与转移。此外,TME中的代谢重编程(如高糖酵解产生的乳酸)进一步维持了Treg的高表达状态,导致单一靶点阻断疗法易产生耐药。因此,双阳性Treg细胞不仅是肿瘤免疫逃逸的核心驱动力,更是预测免疫治疗响应率的关键生物标志物。
清除策略机遇:针对Treg细胞的治疗策略正经历从“单纯阻断信号”向“精准清除细胞”的范式转移,为新一代免疫疗法带来结构性投资机会:
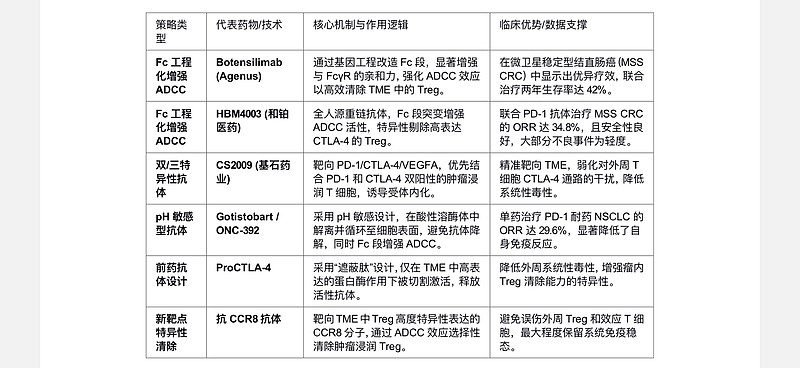
· Fc段工程化改造:传统CTLA-4抗体因毒性限制了剂量,难以有效清除Treg。新一代药物如Agenus的Botensilimab和和铂医药的HBM4003,通过Fc段增强ADCC效应,能精准剔除TME中高表达CTLA-4的Treg细胞,在MSS结直肠癌等难治瘤种中展现出突破性疗效(Botensilimab联合治疗两年生存率达42%)。
· 多特异性抗体协同:双/三特异性抗体成为新风口,如基石药业的CS2009(PD-1/CTLA-4/VEGF)利用亲和力差异,优先结合并清除双阳性肿瘤浸润T细胞,同时降低对外周T细胞的影响,实现了增效减毒。
一、肿瘤微环境Treg细胞表型特征
1. Treg细胞的核心特征与功能重塑
免疫抑制增强:Treg细胞在肿瘤微环境中通过多维机制显著增强其免疫抑制功能,形成肿瘤免疫逃逸的关键屏障:
· 细胞因子分泌与IL-2剥夺:Treg细胞大量分泌IL-10、TGF-β等抑制性细胞因子,直接抑制效应T细胞活性。同时,Treg细胞作为唯一持续高表达CD25(IL-2受体α链)的T细胞亚群,通过“掠夺”微环境中的IL-2,导致效应T细胞因缺乏关键存活因子而凋亡,从而实现被动抑制。
· 接触依赖性抑制:Treg细胞持续高表达CTLA-4(约为效应T细胞的3-5倍),通过高亲和力竞争结合或反式内吞作用(Trans-endocytosis),清除抗原呈递细胞表面的CD80/CD86共刺激分子,从源头阻断效应T细胞的活化信号。
代谢适应重塑:Treg细胞通过代谢重编程适应肿瘤微环境的恶劣条件,并利用代谢产物增强自身功能:
· 乳酸利用与氧化磷酸化:与效应T细胞依赖糖酵解不同,Treg细胞能够利用肿瘤微环境中富集的乳酸作为碳源,通过线粒体氧化磷酸化维持生存,且乳酸代谢可进一步促进其CTLA-4和PD-1的表达,增强免疫抑制能力。
· 氨代谢与尿素循环:在富含氨的肿瘤微环境中,Treg细胞特异性上调精氨琥珀酸裂解酶(ASL)以激活尿素循环进行解毒。同时,通过精胺合酶(SMS)将氨转化为精胺,精胺与PPARγ相互作用增强线粒体功能,使Treg细胞在高氨环境中不仅能存活,还能提升抑制活性。
特异性募集机制:肿瘤细胞通过特定的趋化因子轴主动重塑免疫微环境,实现Treg细胞的定向富集:
· CCL1-CCR8轴:肿瘤细胞分泌趋化因子CCL1,特异性招募表达CCR8受体的Treg细胞迁移至肿瘤部位。
· 高特异性富集:CCR8是肿瘤浸润Treg细胞的高度特异性标志物,在乳腺癌、结直肠癌等多种实体瘤中,高达80%的肿瘤浸润Treg细胞表达CCR8,且主要富集于具有强抑制功能的效应Treg(eTreg)亚群,而外周血Treg细胞极少表达,这为靶向清除肿瘤内Treg提供了精准窗口。
2. Treg细胞的关键表型标记体系
核心转录因子:FOXP3(Forkhead Box P3)是Treg细胞谱系特异性的核心转录因子,对其发育、分化及免疫抑制功能的维持起决定性作用。在肿瘤微环境(TME)中,FOXP3不仅直接调控CTLA-4、CD25等关键效应分子的转录表达,还通过驱动精胺合酶(SMS)的表达促进代谢重编程,使Treg细胞能够利用氨转化为精胺,从而增强氧化磷酸化以适应TME的代谢压力。FOXP3的持续高表达是区分Treg细胞与效应T细胞的最根本特征,其表达水平直接关联Treg的抑制活性稳定性。
关键表面受体:CD25(IL-2受体α链)是Treg细胞表面最显著的标志物之一,呈现持续性高表达状态。Treg细胞通过由CD25、CD122和CD132组成的高亲和力IL-2受体复合物,高效结合并消耗微环境中的IL-2(“IL-2掠夺”机制),导致效应T细胞因缺乏必要的生存因子而无法活化增殖。此外,IL-2信号通过JAK-STAT5通路维持FOXP3的表达及Treg细胞的存活,形成正反馈调节回路。
免疫检查点:CTLA-4(细胞毒性T淋巴细胞相关抗原4)在Treg细胞表面呈组成性高表达,其表达水平约为效应T细胞的3-5倍。Treg细胞利用CTLA-4的高亲和力竞争性结合抗原呈递细胞(APC)表面的CD80/CD86,并通过“反式内吞作用”(Trans-endocytosis)将这些共刺激分子摄入胞内降解,从而阻断效应T细胞活化所需的第二信号。关于PD-1,虽然传统观点认为Treg主要表达CTLA-4,但在TME的高糖酵解和高乳酸环境中,Treg细胞可被诱导表达PD-1,这部分PD-1+CTLA-4+双阳性Treg通常表现出更强的免疫抑制活性和代谢适应性。
肿瘤特异标记:CCR8(趋化因子受体8)已被确认为肿瘤浸润Treg细胞(TI-Treg)的高度特异性标志物。数据显示,在乳酸癌、结直肠癌等多种实体瘤中,高达80%的肿瘤浸润FOXP3+ Treg细胞表达CCR8,而外周血Treg细胞极少表达。CCR8+ Treg细胞主要富集于具有强抑制功能的效应Treg(eTreg)亚群,这使其成为区别于正常组织Treg、实现精准靶向清除而不诱发系统性自身免疫反应的理想靶点。
二、CTLA-4与PD-1表达模式深度解析
1. CTLA-4在Treg中的表达与反式内吞机制
高表达特征:调节性T细胞(Treg)在肿瘤微环境中表现出CTLA-4的持续性高表达特征,其表达水平通常是效应T细胞(Teff)的3-5倍。与Teff仅在活化后短暂表达不同,Treg细胞中的CTLA-4不仅在细胞膜表面表达,还大量存在于胞内储存池中,并通过快速的胞吞和再循环机制在细胞表面与胞内之间动态穿梭。这种独特的表达模式由Treg的核心转录因子FOXP3直接驱动,确保了Treg细胞能够持续发挥免疫监视和抑制功能。
微环境调控:肿瘤微环境(TME)中的代谢因素对Treg细胞CTLA-4的表达具有显著的正向调控作用。研究表明,TME中高浓度的乳酸等代谢产物不仅重塑Treg的代谢适应性,还通过特定的分子机制(如USP39介导的RNA剪接)促进CTLA-4的转录和表达。这种代谢-免疫耦联机制使得Treg细胞在缺氧、酸性的恶劣微环境中仍能维持高水平的CTLA-4表达,从而强化其免疫抑制表型。
反式内吞机制:CTLA-4介导的反式内吞(Trans-endocytosis)是Treg细胞发挥接触依赖性免疫抑制的核心机制。Treg表面的CTLA-4以极高的亲和力结合抗原呈递细胞(APC)表面的共刺激分子CD80和CD86,随后将其从APC表面“剥离”并内吞至Treg胞内进行溶酶体降解。这一过程导致APC表面的CD80/86分子耗竭,使其无法向效应T细胞提供必要的CD28共刺激信号(第二信号),从而从源头上阻断T细胞的活化与增殖。该机制不仅物理性清除了共刺激配体,还通过诱导APC产生抑制性分子(如IDO),进一步放大免疫抑制效应。
2. PD-1在Treg中的诱导性表达与功能争议
表达争议与诱导:学术界关于Treg细胞表面PD-1表达模式的认知经历了显著演变。早期观点倾向于认为Treg细胞主要特征性高表达CTLA-4,而PD-1表达缺失或极低。然而,近期深度研究揭示,在肿瘤微环境(TME)中,Treg细胞的PD-1表达具有显著的诱导性特征,而非固有表达。这种表达模式的高度异质性主要受微环境信号驱动,特别是TME中高糖酵解代谢产生的乳酸积累,被证实是诱导Treg细胞上调PD-1表达的关键代谢信号。这表明PD-1在Treg中的出现并非随机事件,而是细胞适应恶劣微环境的特定反应机制。
代谢适应与稳态:PD-1信号通路在维持Treg细胞的代谢适应性与谱系稳定性方面发挥着核心作用。与效应T细胞中PD-1介导的“耗竭”信号不同,Treg细胞中的PD-1信号对于维持其线粒体功能和代谢稳态至关重要。数据显示,PD-1的缺失会导致Treg细胞发生严重的代谢紊乱,进而破坏其谱系稳定性(Lineage Stability)。PD-1/PD-L1轴通过精细调控糖酵解与氧化磷酸化的平衡,使Treg细胞能够在低氧、营养匮乏的肿瘤微环境中保持存活优势并维持其抑制表型。
抑制增强与耐药:PD-1阳性Treg细胞在功能上表现出更强的免疫抑制活性,这一发现对现有的免疫治疗逻辑提出了挑战。研究指出,PD-1的表达增强了Treg细胞的抑制功能,而非削弱。在临床治疗中,抗PD-1抗体在激活效应T细胞的同时,可能意外地增强了PD-1+ Treg细胞的存活与功能,这种“双向激活”效应被认为是导致部分患者出现免疫治疗耐药甚至超进展(Hyperprogression)的重要机制之一。因此,单纯阻断PD-1可能不足以逆转TME的免疫抑制状态,需结合CTLA-4阻断或代谢干预策略以克服Treg介导的耐药性。
三、双阳性Treg跨肿瘤分布与临床意义
1. 双阳性Treg细胞的跨肿瘤分布图谱
分布概况:CTLA-4⁺PD-1⁺双阳性Treg细胞并非均匀分布于所有组织,而是呈现出显著的肿瘤微环境(TME)富集特征。研究表明,该亚群主要集中在肿瘤浸润区域及肿瘤引流淋巴结中,而在外周血中比例相对较低。这种特异性分布提示双阳性Treg细胞是肿瘤免疫逃逸的关键驱动者,其共表达模式往往伴随着更强的免疫抑制功能和代谢适应性,是连接肿瘤代谢重编程与免疫耐受的核心细胞亚群。
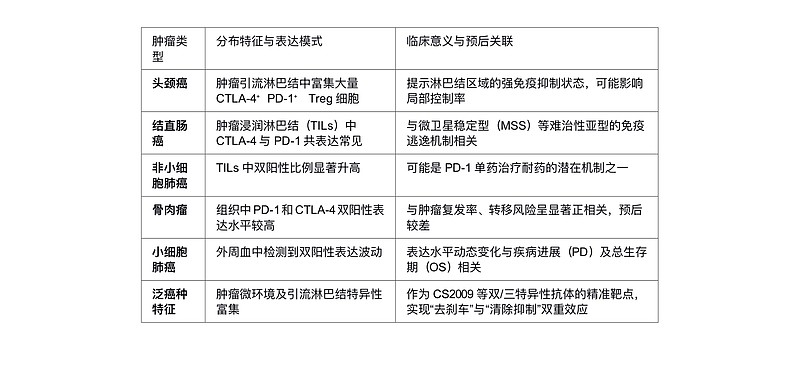
癌种特征:不同肿瘤类型中双阳性Treg细胞的浸润丰度与临床病理特征存在差异。在头颈癌中,肿瘤引流淋巴结内检测到大量CTLA-4⁺PD-1⁺ Treg细胞,这可能解释了该区域强烈的免疫抑制状态。结直肠癌、非小细胞肺癌(NSCLC)、肾细胞癌及黑色素瘤的肿瘤浸润淋巴结(TILs)中,CTLA-4与PD-1的共表达现象尤为普遍,且常与肿瘤的高侵袭性相关。而在骨肉瘤中,双阳性表达水平直接与肿瘤的复发率及转移风险呈正相关,表明其在恶性骨肿瘤进展中扮演重要角色。
临床意义:双阳性Treg细胞的临床价值主要体现在预后预测与精准治疗两个维度。一方面,该亚群的高浸润通常预示着不良预后,如在小细胞肺癌和骨肉瘤患者中,其外周血或组织中的高表达水平与疾病进展(PFS缩短)及转移密切相关。另一方面,作为免疫治疗的新兴靶点,双阳性Treg细胞对常规单靶点抑制剂可能存在耐药性,但对新型多特异性抗体敏感。例如,三特异性抗体CS2009通过优先结合PD-1⁺CTLA-4⁺双阳性细胞,能更精准地清除肿瘤内Treg,同时减少对外周系统的毒性。
分布图谱:
2. 双阳性Treg细胞的功能特质与生存优势
超级抑制特质:CTLA-4⁺PD-1⁺双阳性Treg细胞在肿瘤微环境中表现出“超级抑制者”的表型,其抑制效应T细胞增殖的能力显著强于单阳性亚群。该亚群通过高表达CTLA-4介导“反式内吞”作用,主动剥夺抗原呈递细胞表面的CD80/CD86配体,从源头切断效应T细胞的共刺激信号。同时,PD-1信号通路的激活有助于维持Treg细胞的谱系稳定性及代谢稳态,防止其在炎症环境中转化为效应T细胞,从而保障其持续发挥免疫负调控功能。
代谢生存优势:双阳性Treg细胞展现出卓越的代谢适应性,使其能在低氧、低糖及高乳酸的恶劣肿瘤微环境中存活并扩增。研究显示,该类细胞通过代谢重编程,如利用乳酸作为替代碳源、激活尿素循环以解毒高浓度的氨,从而在与效应T细胞的代谢竞争中占据优势。此外,IL-2信号通路持续激活下游STAT5,上调抗凋亡蛋白Bcl-2的表达,赋予双阳性Treg细胞极强的抗凋亡能力,使其成为肿瘤微环境中寿命最长的免疫抑制细胞群之一。
临床耐药影响:作为免疫治疗后残留Treg的主要形式,双阳性Treg细胞是导致PD-1/CTLA-4单药治疗耐药的关键因素。单一阻断PD-1可能因解除对Treg的抑制而导致其代偿性扩增,进而加剧免疫抑制。临床数据显示,该亚群在肿瘤浸润淋巴结中高度富集,且与不良预后密切相关。针对这一机制,新型双特异性抗体(如CS2009)被设计为优先结合双阳性细胞,通过协同诱导受体内化及增强ADCC效应,实现对这一核心抑制性亚群的精准清除。
四、调控机制与新型免疫治疗策略
1. Treg细胞表达调控网络与分子机制
分子调控网络:肿瘤微环境(TME)中的Treg细胞通过整合微环境信号、胞内转导通路及转录调控,构建了CTLA-4和PD-1共表达的复杂网络。微环境中的关键因子如IL-2、TGF-β及代谢产物(乳酸、低氧)激活JAK/STAT、PI3K/AKT等信号通路,协同驱动免疫检查点分子的表达。CTLA-4在Treg表面的高表达不仅依赖于转录激活,还受胞内储存池与胞膜间内吞循环动态平衡的精密调控。相比之下,PD-1的表达具有更强的环境诱导性,高糖酵解环境及乳酸堆积通过代谢重编程信号显著上调Treg细胞表面的PD-1水平,使其呈现CTLA-4⁺PD-1⁺的双阳性表型,从而在TME中发挥更强的免疫抑制功能。
乳酸剪接调控:肿瘤微环境中富集的乳酸不仅是代谢底物,更是关键的信号分子,通过独特的“乳酸-RNA剪接轴”调控CTLA-4表达。乳酸能够特异性上调RNA剪接因子USP39的水平,进而促进CTLA-4 mRNA前体的有效剪接与成熟,显著增加功能性CTLA-4蛋白的合成。这一机制揭示了代谢微环境直接干预基因转录后修饰的新路径,表明Treg细胞利用肿瘤代谢产物来强化自身的免疫抑制装备,是肿瘤免疫逃逸的重要分子基础。
FOXP3主导作用:FOXP3作为Treg细胞的谱系特异性转录因子,在驱动CTLA-4和PD-1表达中发挥主导作用。FOXP3能直接结合于CTLA-4基因的启动子区域,驱动其组成性高表达,这是Treg细胞区别于效应T细胞的核心特征之一。对于PD-1,FOXP3通过维持Treg细胞的代谢适应性和谱系稳定性,间接支持其在肿瘤微环境中的诱导性表达。此外,FOXP3还与NFAT、AP-1等转录因子形成复合物,协同调控免疫检查点基因的转录活性,确保Treg细胞在复杂微环境中维持稳定的免疫抑制表型。
2. 免疫治疗启示与新型药物策略
单药局限分析:传统免疫检查点抑制剂在临床应用中面临显著挑战。第一代抗CTLA-4抗体(如Ipilimumab)虽然能通过阻断CTLA-4信号激活T细胞,但因无法区分肿瘤微环境(TME)与正常组织中的Treg细胞,常引发严重的免疫相关不良反应(irAEs),导致治疗窗口狭窄。同时,抗PD-1治疗存在耐药风险,研究表明抗PD-1治疗可能导致濒死肿瘤细胞释放氨,进而激活Treg细胞,或因Treg细胞自身表达PD-1而意外增强其存活与功能,反而加剧了免疫抑制。此外,单药阻断容易诱导代偿性免疫抑制通路的激活,使得单一靶点治疗难以在复杂微环境中实现持久应答。
治疗策略演进:针对传统疗法的局限,免疫治疗策略正从单纯的“信号阻断”向“精准清除”演进。新一代药物研发更加重视抗体依赖的细胞介导的细胞毒作用(ADCC),旨在利用TME中Treg细胞高表达CTLA-4、CCR8等分子的特性,通过Fc段工程化改造增强对NK细胞等效应细胞的招募,特异性清除肿瘤浸润Treg细胞。这一策略的核心逻辑在于:通过精准清除TME中的免疫抑制性Treg细胞来打破免疫耐受,同时保留外周Treg功能以维持系统免疫稳态,从而实现“增效减毒”。坂口志文等学者的研究也支持这一观点,认为靶向CTLA-4的核心机制在于清除TME中的Treg,而非单纯的“松刹车”。
新型药物图谱:基于“清除”与“协同”的逻辑,多种新型药物策略已进入临床开发阶段,通过结构优化和机制创新突破传统疗法瓶颈,具体策略如下表所示: