Septerna –SEP631潜在GPCR重磅炸弹?

1. 公司简介
Septerna是一家专注于G蛋白偶联受体(GPCR)药物研发的生物技术公司,总部位于美国加利福尼亚州南旧金山。成立于2019年,2024年上市。该公司由知名生物医药风投机构Third Rock Ventures孵化成立,并吸引了多位GPCR领域的顶尖科学家加入。
科学创始人包括:
Robert Lefkowitz(2012年诺贝尔化学奖得主,GPCR研究先驱)
Brian Kobilka(同为诺奖得主,GPCR结构解析专家)
CEO:Jeffrey Finer,曾任基因泰克高管。
治疗领域:
内分泌、免疫和炎症、代谢性疾病

2. 技术平台
GPCR 对细胞信号传导至关重要,是正常人体生理机能的关键调节因子
G 蛋白偶联受体(GPCR)是细胞膜受体中最大且最多样化的家族,人体几乎每个器官系统都有数百种不同的 GPCR 调节生理过程。它们是药物研发史上成果最丰硕的目标类别,约占所有获美国食品药品监督管理局(FDA)批准药物的三分之一。然而,约 75% 的潜在 GPCR 治疗靶点仍未被药物开发,这为未来的药物研发提供了巨大的未开发机会。
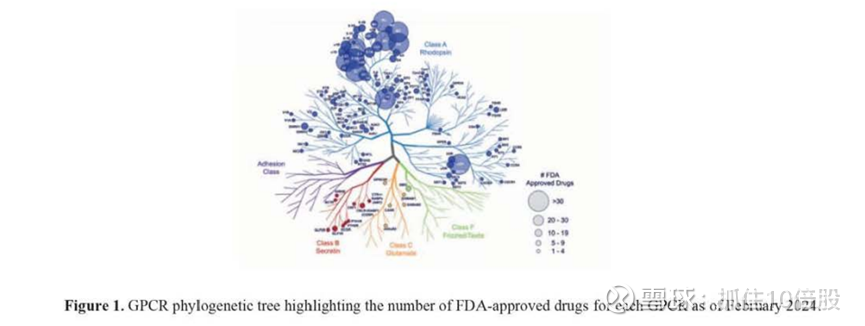
GPCR激活的每一步都涉及到微妙的构象变化,这些变化在历史上很难在细胞外复制。由于无法在细胞外分离出天然功能形式的GPCR蛋白,科学家们无法利用一些最先进的技术,而这些技术在过去十年中已经彻底改变了其他主要靶标类别的药物发现。这一复杂的挑战限制了GPCR药物的发现,特别是新型口服小分子的开发,如肽GPCR激动剂和变构调节剂。迄今为止,药物发现一直高度集中在少数gpcr上。
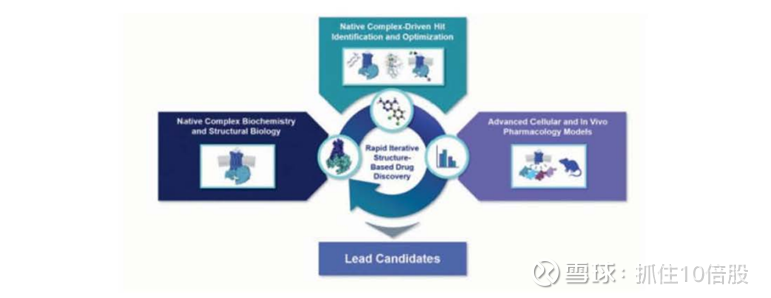
Septerna的核心技术平台名为“Native Complex™”,旨在突破传统GPCR药物开发的限制,通过重组和稳定GPCR天然结构,结合结构生物学、计算建模和高通量筛选,针对以往难以成药的GPCR靶点开发小分子药物。
Septerna 的平台为 GPCR 药物研发开辟新途径
其开发的专有技术,能够在细胞外分离、纯化并重新构建 GPCRs 与配体、转导蛋白和脂质双层的复合物,脂质双层可模拟细胞膜。我们将这些重新构建的组合体称为原生复合物,因为它们在纯化的生化形式中复制了 GPCRs 的天然结构、功能和动态特性。我们的“原生复合平台”由一系列经过优化并整合到专有工业化工作流程中的工具和技术提供支持。这些工作流程包括使用低温电子显微镜快速确定高分辨率 3D GPCR 结构,以及利用超大规模计算筛选和数据库以及利用 DNA 编码库进行生化筛选来筛选数十亿候选分子。
药物发现
平台助力药物候选物的发现与优化
其“原生复合物平台”旨在针对特定的 G 蛋白偶联受体(GPCR),发现经验证受体的新型结合口袋,并探索广泛的药理学,包括激动剂(激活 GPCR 信号传导)、拮抗剂(抑制 GPCR 信号传导)和变构调节剂(通过内源性配体增加或减少 GPCR 激活程度),以不同方式影响 GPCR 信号传导,从而实现理想的治疗效果。
高分辨率的原生复合物 GPCR 结构用于揭示此前未知的结合口袋,为小分子调节 GPCR 的机制提供新的见解,并推动基于结构的药物设计和优化循环快速且反复进行,以发现新的药物候选物。
3. 研发管线
其研发管线主要有 PTH1R激动剂SEP-786、MRGPRX2负变构调节剂SEP-631、TSHR负变构调节剂。
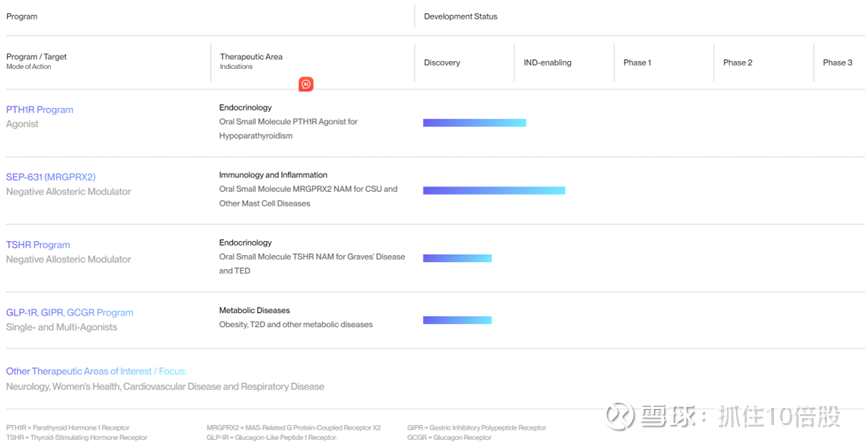
1. PTH1R激动剂(SEP-786)安全性问题1期终止
公司正在开发新型口服小分子甲状旁腺激素1受体(PTH1R)激动剂,用于治疗甲状旁腺功能减退。虽然有针对PTH1R的甲状旁腺功能减退症的肽产品已获批准或正在开发中。但是公司的PTH1R激动剂可以有效和选择性地激活PTH1R, PTH1R是一种高度参与血钙控制的GPCR,有可能在每天一次或每天两次口服给药时实现血清钙和磷酸盐的持续正常化。
2024年第三季度,公司在健康志愿者中启动了SEP-786单次和多次递增剂量(SAD/MAD)的1期临床试验。SEP-786是一种口服小分子PTHIR激动剂候选产品,正在开发用于治疗甲状旁腺功能减退。2025年2月18日,公司宣布决定停止SEP-786的开发,并从PTH1R项目中推进下一代口服小分子PTH1R激动剂。这一决定是在观察到在1期试验的MAD部分出现了两个意想不到的严重(3级)非共轭胆红素升高事件后做出的。
2. MRGPRX2负变构调节剂(SEP-631) -多适应症拓展重磅炸弹潜力
慢性自发性荨麻疹(CSU)是一种全身性炎症性皮肤病,其特征是皮肤上自发和反复出现瘙痒,疼痛的荨麻疹,称为荨麻疹和血管性水肿,或肿胀,在美国影响约150万患者。这些慢性症状通常持续2至5年,可干扰日常生活,包括工作能力,并经常与精神合并症有关,包括抑郁和焦虑。一些CSU患者报告了相关的全身症状,包括头痛和疲劳、喘息、潮红、心悸和胃肠道症状。虽然没有已知的触发因素,但肥大细胞的激活和脱颗粒以及组胺和其他炎症介质的释放导致了CSU的这些衰弱症状。两种典型的途径代表了肥大细胞激活和脱颗粒的主要机制:通过靶向高亲和力IgE受体(FceRI)或IgE本身的抗体通过受体交联激活IgE途径,以及通过MRGPRX2激活IgE非依赖性途径。一旦被激活,肥大细胞就会释放出大量的介质,导致瘙痒、红肿等标志性症状。MRGPRX2在肥大细胞表面高度表达,在肥大细胞活化和脱颗粒过程中起关键作用。这种受体被多种刺激激活,包括神经肽、抗菌肽和某些药物。一旦被激活,MRGPRX2触发信号级联,导致从肥大细胞颗粒中快速释放预先储存的介质,如组胺、蛋白酶和细胞因子。这种脱颗粒过程有助于立即过敏反应和各种炎症条件。MRGPRX2对广泛的配体作出反应的独特能力突出了其在宿主防御机制中的重要性,以及其作为治疗过敏性和炎症性疾病的治疗靶点的潜力。
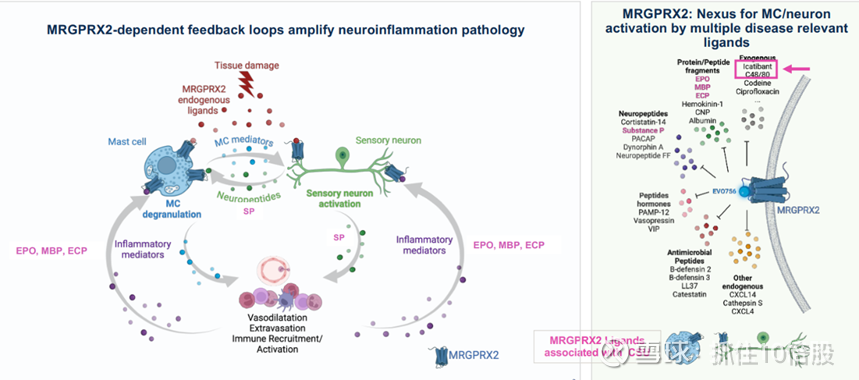
目前的治疗方案及其局限性:
患有CSU的患者最初使用抗组胺药来控制症状;然而,在这种一线环境中,大约37%的患者没有得到充分控制。相当大比例的患者使用抗组胺药后仍有持续症状,这突出了额外治疗方案的必要性。随着对CSU发病机制和肥大细胞作用的深入了解,针对CSU不同驱动因素的新型治疗药物正在开发中。
SEP-631
靶向MRGPRX2的口服小分子可以为CSU患者提供差异化的治疗选择。MRGPRX2 负变构调节剂(NAM)旨在选择性地抑制肥大细胞,将广泛免疫抑制的风险降至最低,这可能与其他机制方法一起观察到,要么根除肥大细胞,要么抑制多种免疫细胞类
型。我们相信选择性肥大细胞抑制剂有潜力成为更安全的治疗选择,可用于单药治疗和联合治疗。有了我们的NAM,我们相信我们可能能够普遍阻断所有内源性MRGPRX2激动剂,即使在高浓度MRGPRX2激动剂存在的情况下也能阻止MRGPRX2的激活。我们相信,结合这些特点,我们的NAM有可能控制患者的症状并防止疾病发作。我们正在开发SEP-631,最初用于治疗CSU,因为我们相信这可能为临床概念验证提供有效途径。由于抗组胺难治性患者很少有口服治疗方案,因此CSU仍有大量未满足的需求。由于多种疾病是由活化的肥大细胞驱动的,我们相信有机会扩展到多个治疗领域的适应症,如过敏性哮喘、特应性皮炎、间质性膀胱炎、偏头痛和结节性痒疹。
临床前活性:
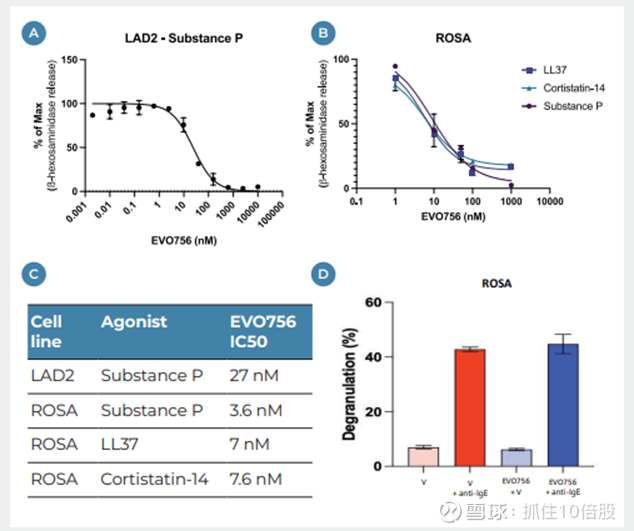
SEP-631在皮质抑素-14 (IC50 1.6 nM)刺激下,可有效阻断HEK293细胞内MRGPRX2过表达的细胞内信号通路的激活。使用不同浓度的SEP-631和不同浓度的皮质激素-14的基质进行的实验显示,SEP-631对最大激动剂效应有很强的抑制作用,我们认为这表明SEP-631有可能成为一种NAM,当与MRGPRX2结合时,无法被过量的MRGPRX2 激动剂所取代。
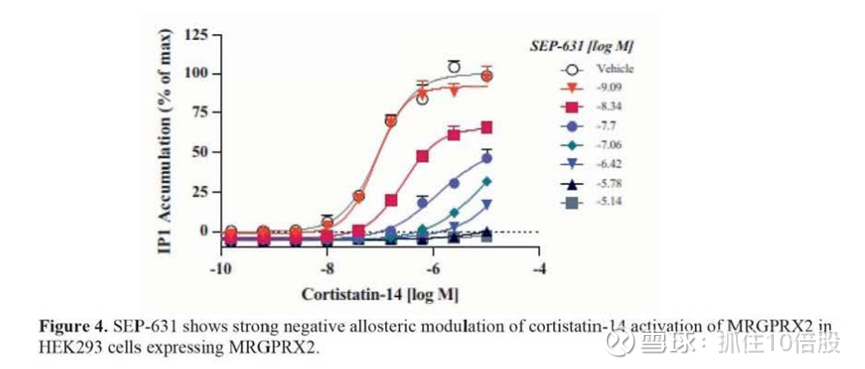
SEP-631在几种临床相关的内源性MRGPRX2激动剂的激活下,可以阻断表达MRGPRX2的HEK293细胞中IP1的积累,表明其抑制作用与激活激动剂无关。
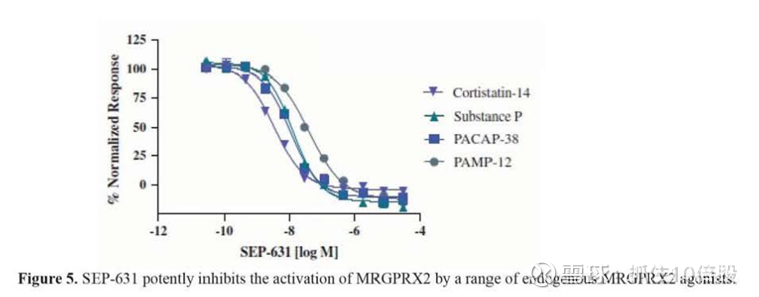
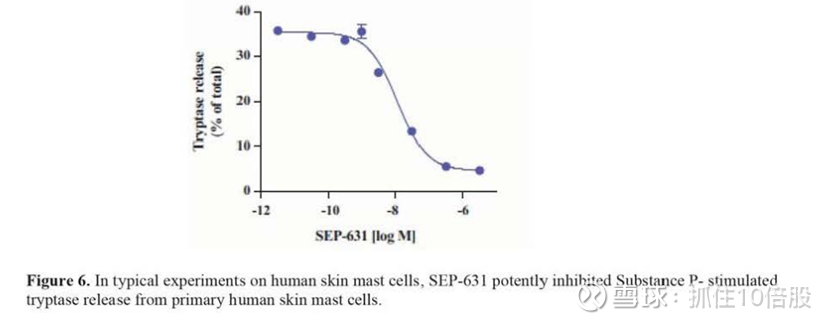
在不同的体外肥大细胞脱颗粒模型中,SEP-631被证明是LAD2细胞(IC50 2.3 nM)和原代人脐血来源肥大细胞(IC50 0.72 nM)的激活和脱颗粒的有效抑制剂。在典型的人皮肤原代肥大细胞实验中,SEP-631完全有效地抑制由EC90浓度P物质(IC50=12 nM)触发的胰蛋白酶释放。
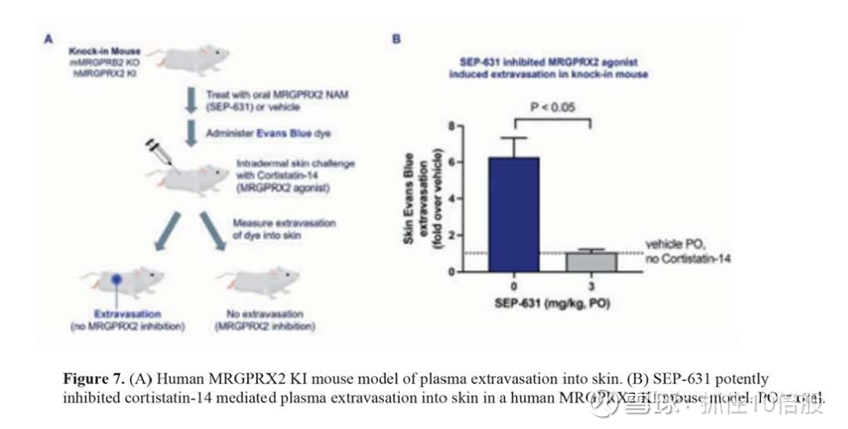
与其他第三方MRGPRX2抑制剂相比,SEP-631的一个关键特征是其靶标停留时间长或抑制速率慢,这意味着受体-配体复合物解离和受体再次激活需要很长时间。采用了两种实验方法来确定受体-配体复合物的半衰期:放射性配体结合实验和一个表面等离子体共振研究分别证明了124分钟(标准偏差为20分钟)和50分钟的长半衰期。受体配体的长靶停留时间被认为对延长药物在体内的作用具有潜在的优势,这已被证明可以转化为增强的临床活性。为了在体内表征SEP-631,我们开发了一种转基因敲入(KI)小鼠模型,其中内源性小鼠MRGPRB2受体的编码区被人类MRGPRX2受体取代,因为小鼠和人类同源物之间具有低序列同源性。在这个模型中,MRGPRX2激动剂配体,如P物质或皮质抑素-14,注射到皮肤后会刺激血浆外渗或水肿。外渗可以通过Evans蓝染料从循环重新分布到皮肤组织来量化。在MRGPRX2 KI小鼠模型中,在皮质抑素-14皮肤刺激前口服SEP-631可有效抑制皮肤外渗,显示口服3mg /kg剂量可完全阻断皮肤肥大细胞脱颗粒。
临床前研究支持SEP-6的临床开发
确定了SEP-631在多个物种中的临床前药物代谢和PK谱,以支持人类PK预测。SEP-631具有高度口服生物利用度的潜力,清除率低,预计半衰期与每日一次口服剂量一致。迄今为止进行的体外和体内安全性研究表明,SEP-631具有良好的耐受性。在大鼠和狗的28天重复口服GLP毒理学研究中,SEP-631的耐受性一般良好,与人类有效剂量下预计的最大暴露量相比有很大的安全边际。
SEP-631的临床开发计划和现状
我们计划首先开发SEP-631用于抗组胺难治性CSU患者。我们预计将于2025年3季度在健康志愿者中进行1期临床试验,以评估安全性、耐受性、PK和PD。除了CSU,我们正在评估一系列其他适应症,其中肥大细胞异常活化已被证明是疾病病理生物学的核心。肥大细胞激活驱动多种流行疾病,包括过敏性哮喘、特应性皮炎、间质性膀胱炎、偏头痛和结节性瘙痒,我们相信SEP-631可以为这些患者群体提供一种新的口服治疗选择。我们计划探索这些适应症作为潜在的未来临床开发机会。
分子结构:
公司尚未披露具体分子结构。目前仅披露专利(WO 2024/226914 A2)
部分结构:
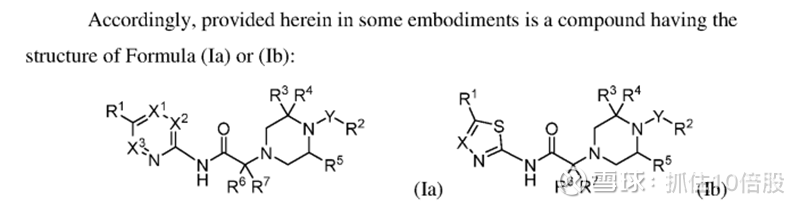
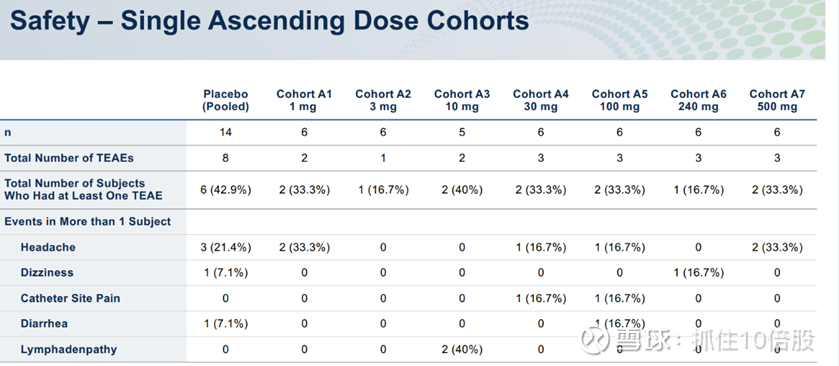
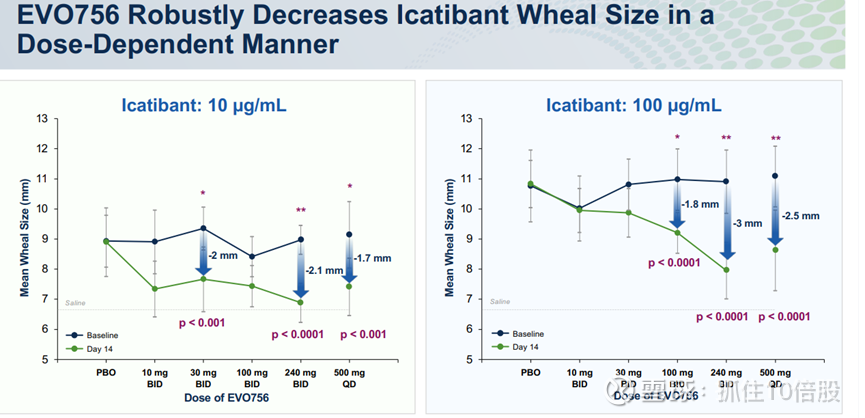
当前该靶点进展最快的为evommune的分子evo756,该分子未披露结构。目前已经完成一期临床,其在10-500mg剂量下安全性良好,支持其进一步的临床试验。
但是SEP-631在LAD2细胞的IC50 为2.3 nM,evo756在LAD2细胞的IC50为27nm。效力约为10倍。当然这仅仅是一个细胞系的数值,并不能比较两个分子优劣。
3.TSHR负变构调节剂(SP-1351)-开发早期先导优化
格雷夫斯病(Graves' disease)是最常见的自身免疫性疾病之一,影响了美国200多万患者,也是甲状腺功能亢进主要原因。在格雷夫斯病中,身体产生自身抗体,结合并激活甲状腺细胞上的TSHR。这些自身抗体刺激甲状腺产生过多的甲状腺激素,导致甲状腺功能亢进。甲状腺激素影响许多身体系统,因此格雷夫斯病的症状可以是广泛的。格雷夫斯病的常见症状包括焦虑、易怒、颤抖、热敏、体重减轻、心跳加快或不规则以及睡眠障碍。虽然格雷夫斯病可能影响任何人,但在女性和40岁以下的人群中更为常见。
TED是一种相关但不同的威胁视力的自身免疫性疾病,在大约50%的Graves病患者中发生。在TED中,自身抗体结合并激活眼后眼眶成纤维细胞上的TSHR,从而导致炎症、眼眶脂肪扩张和纤维化。TED是一种进行性疾病,早期诊断和治疗对于防止恶化和严重的眼睛损伤非常重要,包括眼球突出(眼睛凸起)、斜视(眼睛不对准)和复视(视力模糊或重影)。
目前的治疗方案及其局限性
在过去的70年里,格雷夫斯病最常见的治疗方法基本保持不变,包括抗甲状腺药物,如甲巯咪唑和丙硫尿嘧啶,旨在降低甲状腺产生激素的数量或阻断甲状腺激素对身体的影响,旨在破坏过度活跃的甲状腺细胞的放射性碘疗法,以及切除全部或部分甲状腺的甲状腺切除术。许多患者在抗甲状腺药物治疗后疾病复发率很高,在消融和甲状腺切除术后出现终身甲状腺功能减退。此外,这些治疗方案最初可能会解决潜在的症状,但它们不能改善疾病,也不能阻止疾病进展。目前对TED的治疗取决于疾病的严重程度,旨在帮助控制症状和减缓疾病进展。对于轻度TED患者,改变生活方式和非处方药,如人工泪液和硒补充剂,可能有助于缓解干眼症。对于严重的TED,类固醇和/或眼科手术,如眼眶减压,可以考虑。从历史上看,患者必须与TED一起生活,直到炎症消退,之后他们通常会留下永久性和视力受损的后果,可能需要多次手术,这些手术并不能完全使患者恢复到疾病前的状态。
口服TSHR NAMs的发现和临床前活性
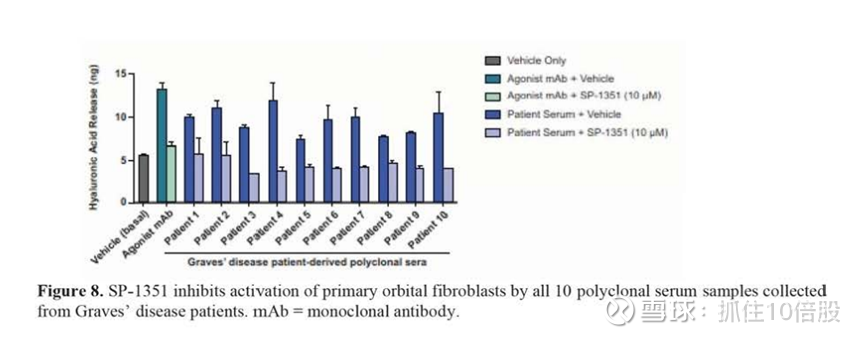
我们已经使用Native Complex Platform鉴定了多种可处理的口服小分子TSHR NAMs的化学系列。利用TSHR NAMs进行的分子药理学研究表明,多个化合物系列具有高效能和理想的药物特性。在表达人类TSHR的细胞中,从格雷夫斯病患者分离的自身抗体激活的cAMP信号被几种先导化合物显著抑制。此外,我们的化合物在抑制TSHR方面表现出对其他gpcr的高选择性。Graves病和TED的有效治疗需要广泛抑制患者自身抗体,这些抗体通常具有高亲和力,并且在活动性疾病期间以高滴度存在。此外,这些自身抗体可能结合到TSHR的大细胞外结构域的不同位点。我们相信TSHR NAM具有优化的药理学特征,可以完全阻断所有患者自身抗体的活性。为了证明我们的TSHR NAM可以完全抑制多种患者自身抗体,我们评估了我们的一种小分子TSHR NAMs (SP-1351)对应用于TED患者眼眶成纤维细胞的Graves病患者源性多克隆血清的活性。血清对成纤维细胞的激活是通过定量细胞产生的透明质酸来测定的。SP-1351能够抑制10个多克隆血清样品中的10个样品的活性,每个样品来自不同的Graves病患者。这一结果表明,我们的TSHR抗体对格雷夫斯病患者中发现的多种多克隆自身抗体具有广泛的抑制活性。
为了表征这些口服TSHR NAMs对体内疾病表现的影响,我们建立了甲亢小鼠翻译模型。长期使用Graves病患者源性TSHR激活抗体治疗的小鼠出现与Graves病患者相似的多种表现,包括血浆甲状腺激素T4水平升高、甲状腺重量增加和甲状腺突出。重复口服SP-1351治疗一周后,其中一些表现显示出逆转的迹象,包括甲状腺激素T4水平正常化,甲状腺重量减少,前列腺增生减少。
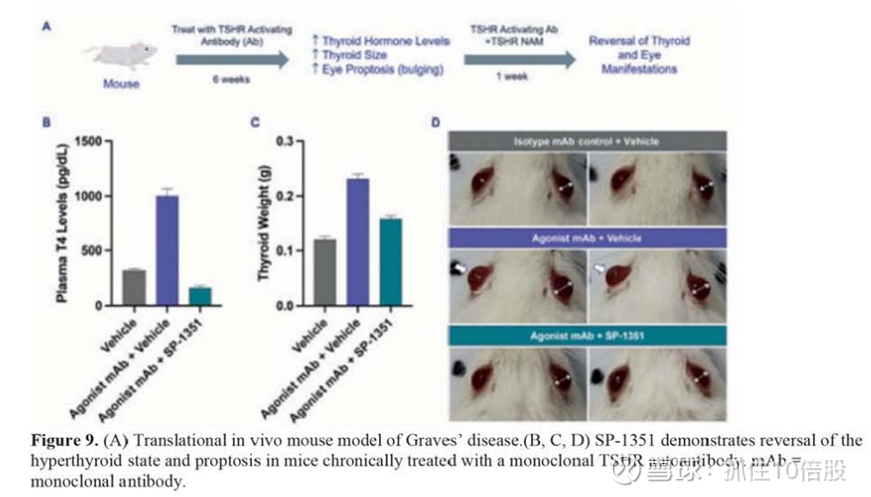
在同一小鼠模型中,评估对甲状腺组织的影响。Graves病患者甲状腺的特点是滤泡增生和/或肥大,细胞内胶体滴,滤泡胶体减少和扇形,血管和淋巴细胞浸润增加,所有这些在我们的小鼠疾病模型中都表现出来。口服SP-1351后,我们观察到滤泡肥大和胶体滴明显减少。
开发计划
我们将继续优化多种早期口服小分子TSHR NAMs,目标是推进先导化合物的选择,使其成为IND研究的开发候选物。在我们的临床前研究中,我们在一种新型Graves病小鼠模型中发现了多个TSHR NAMs,这些NAMs显示出逆转甲状腺功能亢进和增生的能力,并且在使用原代人细胞的基于细胞的检测中抑制了多种Graves病患者TSHR自身抗体。我们打算继续进行TSHR NAM项目的临床开发,以治疗Graves病和TED。
4.财务及估值
2025年5月14日,Septerna和诺和诺德(Novo Nordisk)宣布了一项全球合作协议,将发现、开发和商业化基于特定分子靶点的多种潜在口服小分子药物,用于治疗肥胖、2型糖尿病和其他心脏代谢疾病。两家公司最初将同时启动四个研发项目,针对一种或多种GPCR靶点(包括GLP-1、GIP和胰高血糖素受体)进行小分子治疗。根据协议条款,Septerna有资格从诺和诺德获得约22亿美元的预付款以及研发和商业里程碑付款。这包括超过2亿美元的前期和近期里程碑付款。
截至2025年3月31日,现金、现金等价物和有价证券总计3.982亿美元。若包括诺和诺德2亿美元首付。现金预计达到6亿美元。截止2025年6月15日,公司市值为4.5亿美元。
GPCR一直是十分重要的药物靶点,但是还有众多GPCR靶点未被开发。作为一种跨膜蛋白,其结构研究一直比较困难,GPCR结构的动态变化及复杂的生物机制使得药物的理性设计十分困难。同时很难在体外进行GPCR蛋白的表达和纯化,同时保留其生物特性,导致体外筛选困难。近年来随着结构生物学和计算机模拟技术的发展,GPCR药物发现重新吸引人们的关注。Septerna的“Native Complex”平台有望创新GPCR药物发现。