中国创新药M701获FDA临床试验批准,恶性胸腔积液治疗迈向国际化新阶段

来源:药渡Daily
近日,武汉友芝友生物制药股份有限公司宣布,其自主研发的注射用重组抗EpCAM和CD3人鼠嵌合双特异性抗体M701,已于2026年1月31日正式获得美国食品药物管理局临床试验申请批准。这意味着该药物针对恶性胸腔积液的临床研发正式进入国际化阶段,有望为全球患者提供一种新的局部治疗选择。

---------------来源:友芝友生物公告
恶性胸腔积液是晚期肺癌、乳腺癌等多种实体肿瘤常见的严重并发症,患者常伴有胸闷、呼吸困难等症状,生活质量严重下降,且预后通常较差。目前临床上多以胸腔穿刺引流、局部化疗等姑息性治疗为主,缺乏安全、长效且可重复使用的针对性药物,治疗空白亟待填补。
M701作为一种双特异性抗体药物,其独特之处在于可同时靶向上皮细胞黏附分子和T细胞表面的CD3抗原。EpCAM在多种上皮源性肿瘤细胞表面高表达,而CD3是T细胞免疫应答的关键受体。M701通过“识别肿瘤细胞+激活T细胞”的双重机制,在胸腔局部形成精准免疫杀伤效应,从而实现针对恶性胸腔积液的靶向治疗。
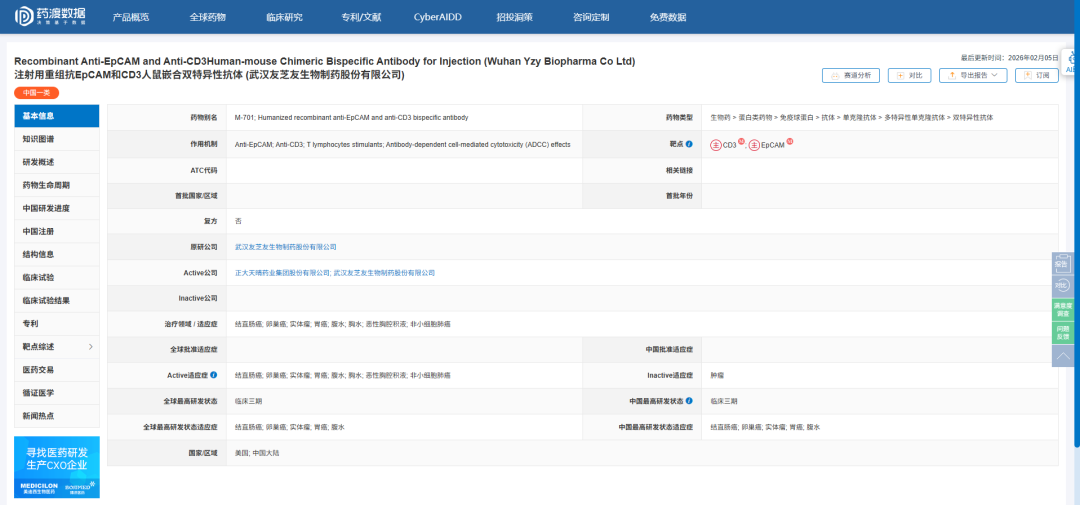
根据公司此前在2025年欧洲肿瘤内科学会会议上公布的II期临床中期数据,M701在治疗非小细胞肺癌所致恶性胸水方面已显示出积极疗效。截至2025年3月,接受M701治疗的患者中位无穿刺生存时间达到130天,优于对照组的85天,且在驱动基因阴性或既往接受过胸腔内化疗的患者中获益更为显著。安全性方面,M701治疗相关不良事件发生率较低,提示其在胸腔灌注给药方式下具备良好的耐受性。
本次FDA批准的是一项开放标签、多中心Ib/II期临床研究,旨在进一步评估M701经胸腔内给药治疗晚期上皮性肿瘤所致恶性胸腔积液的安全性、有效性、药代动力学及免疫原性等关键指标。该研究将在国际多中心开展,标志着M701从中国本土临床走向全球系统评估的重要跨越。
从国内临床探索到获得FDA IND批准,M701的推进路径不仅体现了友芝友生物在肿瘤并发症治疗领域的持续深耕,也展现了中国创新药在国际化临床开发中的积极进展。随着该药物进入国际临床阶段,未来有望推动恶性胸腔积液从“反复穿刺、对症处理”的被动模式,转向“药物干预、精准治疗”的主动管理新阶段,为患者带来更长的无症状生存期、更稳定的生活质量和更有尊严的治疗体验。
在全球肿瘤治疗日益强调精准与个体化的今天,M701的国际化临床开发不仅是一次企业层面的突破,更是中国创新药走向世界、参与全球疾病治疗进程的生动缩影。我们期待在不久的将来,该药物能为更多受恶性胸腔积液困扰的患者带来实质性的治疗希望。
*声明:本文仅是介绍医药疾病领域研究进展或简述研究概况或分享医药相关讯息,并非也不会进行治疗或诊断方案推荐,也不对相关投资构成任何建议。内容如有疏漏,欢迎沟通指出!