亚盛-多发性骨髓瘤MM

MM 是“骨髓里的浆细胞癌”,会分泌单克隆免疫球蛋白(M 蛋白/轻链),逐步造成骨、肾、造血与免疫功能损伤。它与MDS的区别是,MDS是骨髓里的造血干/祖细胞克隆异常 引起无效造血 从而导致贫血、中性粒细胞、血小板减少。两者都可“贫血+乏力”,但 MM 常伴 M 蛋白/骨痛/肾损。
MM的四类损伤:高钙(C)、肾损害(R)、贫血(A)、骨病变/疼痛或骨折(B)
2025 年美国估计新发约 36110 例,死亡约 12030 例,存量20万人,大多数患者诊断时 ≥65 岁,平均诊断年龄约 69 岁。移植8300人。移植者5年的OS率80%,不移植者D-Rd方案的 OS 90月。
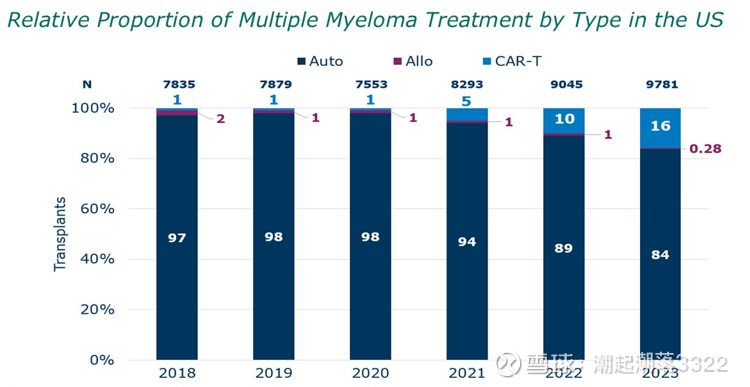
初诊分层:确认CRAB/SLiM标准、ISS/R-ISS、FISH/NGS(del(17p)、t(4;14)、t(14;16)、1q增多等高危标志),评估脏器功能与脆弱度。
移植
MM首选自体移植(ASCT),而非常规做异基因移植(allo-HCT)。原因是自体能在低治疗相关死亡率(TRM)和无GVHD的前提下,稳定带来更深缓解与更长首个无进展期;异体虽有“移植物抗骨髓瘤(GvM)”效应、复发率更低的潜力,但TRM/GVHD代价高且未证实总体生存优势。异基因移植(allo-HCT)“死亡率高”的核心原因在于免疫学并发症(尤其移植物抗宿主病,GVHD)与长期免疫抑制引发的感染叠加,再加上预处理方案的器官毒性、移植物失败、肺部并发症(IPS/DAH/晚期非感染性肺病)等,共同导致非复发死亡(NRM/TRM)。AML 做异基因移植,2年NRM约 11%/16%/27%/33%(由低→高风险),AML必须用异体的原因是可以降低复发。
MM的治疗历史
MM在1996年以前是以化疗+激素,MP(美法仑+泼尼松)为治疗方案,不做移植,当时的OS是3年。1996-2003年确立了耐受人群的自体移植作为一线方案。
1996:法国 IFM 研究显示高剂量美法仑+ASCT 相比常规化疗显著延长无事件生存和总生存 首次建立优势。
2003:英国 MRC Myeloma VII再次证实高剂量治疗+ASCT 优于标准剂量方案 → 巩固为“可移植患者”的一线标准策略。
这之后,市场根据患者身体条件分为两条线,一条是移植者的治疗方案,一条是不移植者。
移植者的治疗路径如下:
1997–2002,以VAD(长春新碱/多柔比星/地塞米松)或TD(沙利度胺/地塞米松)作诱导,进入ASCT,巩固治疗没有标准方案。
2010年,确立了VTD(硼替佐米+沙利度胺+地塞米松)三联方案成为主流诱导,诱导后 CR/近CR:31% vs 11%。
2012年,来那度胺一线治疗坐实移植后维持治疗,PFS 41 vs 23 个月,4 年 OS 相近。
2015年,VRd (硼替佐米+来那度胺+地塞米松)三联确立移植者治疗的诱导+维持(来那度胺)一线方案,PFS:43 vs 30 个月,OS:75 vs 64 个月。
2017年IFM/DFCI 2009临床:早期ASCT vs VRd不移植在现代三联背景下,VRd+ASCT较单纯VRd(不做 upfront 移植)显著延长PFS,OS无差异;巩固与1年维持纳入路径。此后VRd诱导→ASCT→VRd巩固→来那度胺维持成为标准范式。
2019–2020,CASSIOPEIA:D-VTd优于VTd,PFS,83.7 个月 vs 52.8 个月。欧盟委员会于2020-01-20批准 D-VTd 用于可移植NDMM。2019-09-26,FDA批准达雷妥尤单抗(IV)+VTd用于可移植NDMM的诱导+巩固。
2021–2023,FORTE 显示KRd诱导+ASCT并以KRd或Len维持可带来更深缓解/更长PFS,3年PFS 75% vs 65%,HR 0.64。ATLAS三期临床验证,KRd 维持 vs R 单药:中位PFS 59.1 vs 41.4个月;HR 0.51。KRd成为部分中心的高强度诱导选择。卡非佐米是第二代选择性蛋白酶体抑制剂替代硼替佐米的。
2022年DETERMINATION(NEJM):VRd+ASCT再次证明PFS优势 在Len维持直至进展,早期ASCT仍显著延长mPFS 67.5 vs 46.2 月(5 年 OS 80.7% vs 79.2%),进一步巩固“三联/四联+ASCT+维持”的一线总框架。≥3 级治疗相关不良事件 94.2% vs 78.2%。
2024年四联方案(D-VRd)确立,PERSEUS,4年PFS率:D-VRd 84.3% vs VRd 67.7%。FDA于7月30日批准 D-VRd用于可移植NDMM 的诱导+巩固并配套(达雷妥尤单抗+来那度胺)或 来那度胺维持。标准危多为来那度胺单药维持,高危更倾向双药维持(硼替佐米+来那度胺)。在移植后维持,达雷妥尤单抗+来那度胺(D-R)在 NCCN 属于 (其他推荐)/Category 2A。
不移植者的治疗路径如下:
2006–2007,美法仑+泼尼松+沙利度胺(MPT)优于MP,3年OS:MPT 80% vs MP 64%。
2008 ,VISTA研究证实硼替佐米+MP(VMP)显著优于MP,确立VMP为不移植一线强力方案(尤其欧洲),中位OS:56.4个月(VMP)vs 43.1个月(MP)。
2010,VMPT(硼替佐米+美法仑+泼尼松+沙利度胺)→VT (硼替佐米+沙利度胺)维持 vs VMP显示PFS优势。mPFS 35.3 vs 24.8 月,5年OS 61% vs 51%。采用受毒性限制,外周神经病变过多。
2012,MPR-R(诱导MPR(美法仑+泼尼松+来那度胺)→来那度胺维持)显著延长PFS MM-015证实MPR-R优于MPR/MP,奠定维持理念。中位PFS:31个月 vs 14个月,OS:随访时未见显著差异(受后续治疗影响大)。
2014,FIRST试验证实持续来那度胺+地塞米松(Rd)优于MPT(含OS获益)把美国不移植一线从含沙利度胺的 MPT切换到连续Rd作为主路径,确立了一直给药到进展。PFS:25.5个月(Rd-持续) vs 20.7个月(Rd-18) vs 21.2个月(MPT),中位OS 59.1个月(Rd-持续) vs 49.1个月(MPT)。
2016/2017,SWOG S0777:VRd > Rd,无意立即移植人群(多数为不移植/延期移植)中,VRd较Rd显著延长PFS/OS,催生了VRd-lite(周给硼替佐米、降强度)在适度体能人群中的一线应用。中位PFS:43.0 vs 30.0个月,中位OS:75.0 vs 64.0个月。
2018,ALCYONE:D-VMP成为不移植新标准之一 达雷妥尤单抗+D-VMP显著优于VMP并获批,成为欧洲/多地区不移植一线主力组合。PFS 36.4 vs 19.3 月,OS 83.0 vs 53.6 月。
2019,MAIA:D-Rd的CD38三联范式D-Rd较Rd,mPFS进一步拉大到约61.9 vs 34.4个月,OS约90.3个月对64.1个月,D-Rd显著提高MRD阴性率(32% vs 11%),随后被各大指南列为不移植首选。2020 Dara 皮下制剂(Faspro)获批,D-Rd/D-VMP给药便利性大幅提升,加速其一线渗透。
2021,EHA-ESMO 指南:D-Rd与D-VMP并列不移植标准 欧洲权威路径把两套CD38三联都写入一线首选。
2023,VRd-lite(周给 V + 低强度)在老年适度体能者的可行优选(实践共识增强) 多项回顾/队列与专家共识推动周给V+降强度成为“既要疗效也要耐受”的现实方案。中位 PFS 35.1 月,外周神经病变仅 1 例 ≥3 级。
2024,Isa-VRd(异达妥+VRd)在不移植人群获FDA批准 IMROZ显示与VRd相比PFS显著改善,FDA于2024-09-20批准Isa-VRd用于不适合自体移植一线,60个月PFS率:Isa-VRd 63.2% vs VRd 45.2%,静脉给药,赛诺菲。在不移植人群,D-Rd与Isa-VRd有竞争了,前者皮下,后者静脉。
2025,CEPHEUS公布:D-VRd在“不移植或延期移植”人群优于VRd(终末PFS分析) 进一步巩固抗CD38+三联/四联在NTE中的优势格局,目前还没获批不移植的四联方案。D-VRd对照VRd,PFS:HR 0.57;中位 PFS 未达到 vs 52.6 月;54 月 PFS 率 68.1% vs 49.5%;MRD⁻(10⁻⁵) 60.9% vs 39.4%。D-VRd 与 D-Rd(MAIA)构成两条主流达雷妥尤单抗方案;是否加 V(VRd→D-VRd)更多取决于体能/周给可行性/神经毒性顾虑与深度优先目标。 多国指南与综述都建议按 IMWG 脆弱度评分。这类人群往往更能长期承受 D-Rd(毒性与就诊负担更低),而 D-VRd/VRd 的累积毒性与管理成本更高。
MM联用的四款药:来那度胺、达雷妥尤单抗、硼替佐米、地塞米松。其中出来两款百亿美元的大药,来那度胺和达雷妥尤单抗,现在这俩药依然在移植者和不移植者的一线方案中占据主要位置,来那度胺在2021年就专利到期,峰值在121亿。达雷妥尤单抗今年140亿美元,还有20%的增速。如果两药专利都不到期,今年估计合计销售破300亿美元了。由于OS长,这俩药获批适应症时以PFS为主要终点。
四药联用的治疗逻辑
MM的病因是多步演化的结果:1.遗传启动 + 进化:早期常见IGH易位(如 t(11;14)、t(4;14))或高倍体;随后不断积累次级突变(RAS/MAPK、MYC、1q扩增、TP53缺失等),形成克隆与亚克隆。2.骨髓微环境喂养:基质细胞/巨噬/内皮细胞分泌 IL-6、BAFF、APRIL 等,让瘤细胞更抗凋亡;同时驱动溶骨(RANKL↑、DKK1↑)。3.免疫逃逸:T细胞疲惫、抑制性细胞(Tregs/MDSCs)增多,瘤细胞表面 CD38 高表达 等,使其躲过免疫清除。
MM很少有专门靶向药,因为大部分不是靠一个必需突变活着,堵一个口,旁路就起。治疗压力一来,主克隆/副克隆此起彼伏,易耐药。很多异常是“转录/表观层面”改写(如t(4;14)的NSD2/MMSET),药物难以精确命中。与其盯某个罕见突变,不如打它的共性依赖(蛋白应激、免疫清除、基因调控网络)。这也是PI+IMiD+抗CD38成为主轴的生物学理由。
达雷妥尤单抗D(抗CD38):对准免疫逃逸,盯着CD38把髓浆细胞直接清掉(补体、ADCC/ADCP等),还能整理免疫环境,更容易达到更深缓解/MRD阴性。加上去普遍能显著拉长PFS,而且耐受好。靶点普适:CD38 在恶性浆细胞普遍高表达,无需分子分型挑人,适配面广。毒性谱互补:DARA 本身药代/相互作用少(单抗),主要问题是早期输注反应与感染风险,相对不叠加外周神经毒/心毒,便于和 IMiD/PI协同。
来那度胺R(IMiD):来那度胺(Len)属于第二代IMiD,取代了沙利度胺(Thal),原因是疗效证据更强、可长期给药、外周神经病变更少。一边抑制骨髓瘤细胞增殖,一边调动T细胞/NK细胞,和抗CD38抗体配合更强,显著增强达雷妥尤单抗的ADCC效应(抗体依赖的细胞介导细胞毒作用)。可口服、能长期吃,、可按肾功能降阶。这让“持续治疗/维持”在老年与合并症人群成为现实,长期门诊管理成本—获益比优。
硼替佐米V(PI):抑制蛋白酶体,杀伤快、对肾功能不好、t(4;14)/del(17p)等高危患者尤其重要。缺点是打针+周给、外周神经病变和带状疱疹风险。
地塞米松d(激素):本身就杀瘤,能显著增强上面三种药的效果;还减少单抗输注反应、缓解疼痛炎症、改善症状。便宜、可快速见效、可灵活减量,几乎所有方案都离不开它。
为什么更容易先拿掉硼替佐米?很多不移植/延期移植患者,用D+R+d(三联)已经能达到非常强的疗效与持久度;这时再加V的边际收益相对变小,但门诊负担和神经毒性却明显上升。V更像前期快速降负荷的工具,等肿瘤压下去后,保留D与R长期控场,把V撤掉很常见。
来那度胺R的作用是长期控场,达雷妥尤单抗D的作用把深度做出来,地塞米松d的作用既增敏又快速缓解症状。硼替佐米V的作用起始快速削负荷,尤其救急。
基于分子标志物
t(11;14) → BCL-2更依赖 → 维奈克拉,维奈克拉在这群体反应更好;但在未筛选人群里曾出现早期死亡增加(BELLINI)。同类三期(CANOVA,VenDex对PomDex)PFS未达显著,但在生物学选择好的亚组仍有信号。NCCN把Ven+dex(±达雷/±PI)列为t(11;14)复发MM的治疗选项。
BRAF V600E(约3–10%)→ BRAF/MEK抑制,个案与小系列显示达拉非尼+曲美替尼在这小群体可奏效(多为“个例/篮子研究”层级,非标准一线)。
t(4;14):理论上FGFR3/NSD2驱动,但FGFR抑制剂临床转化有限;反而是蛋白酶体抑制剂(如硼替佐米)显著改善了这群体的长期结局,成了“功能性精准”的代表。
针对不移植人群的CEPHEUS的高危亚组(del17p、t(4;14)、t(14;16)),52 例(D-VRd 27;VRd 25),中位PFS 39.8 vs 31.7 月 与 HR 0.88,未达显著,官方解释是:样本量小、D-VRd 组治疗周期更短导致缺失的 MRD 随访样本更多,对结果有影响”,这部分人群占25%。
针对移植人群的PERSEUS,高危= del17p / t(4;14) / t(14;16)):D-VRd 组:76 例;VRd 组:78 例(合计 154 例),PFS HR = 0.59(95%CI 0.36–0.99)
靶点抗原表达:BCMA / GPRC5D(多数MM表达BCMA)。BCMA定向:CAR-T(Cilta-cel/Carvykti;Ide-cel/Abecma),或双特异抗体(Teclistamab/Elranatamab);GPRC5D定向:Talquetamab。多线复发或三类耐药:优先考虑BCMA定向(CAR-T或双抗),或GPRC5D双抗作为非交叉机制出路。
硼替佐米
武田。2003年FDA获批MM的经治后进展市场,2008年批准初治MM一线(经典VMP:硼替佐米+美法仑+泼尼松),2012-01-23:批准皮下给药适用于全部已批准适应症,2013年全球销售峰值约26亿美元。后面开始下滑是因为一线不移植人群从“含美法仑方案”切到连续来那度胺+地塞米松(Rd)路径。达雷妥尤单抗进入前线后,也以D+Rd作为不适合移植人群的主要方案,硼替佐米在不移植人群被剔除方案了,2025年的临床又加上了,能获得更好的PFS和MRD⁻。之所以加加减减是因为加上硼替佐米疗效好,但毒性变大不小,SWOG S0777 中 VRd 的≥3 级神经毒性明显高于 Rd(24% vs 5% );很多患者因此提前停 V。并且硼替佐米的给药特别麻烦,比如诱导期打8个周期,21天一轮,每个周期打第1/4/8/11天。2022年专利到期。
来那度胺
BMS的。2006年获批MM复发难治3亿美元,2012年移植者和非移植者一线维持治疗,销售37亿美元,2014年2月获批非移植人群一线从诱导开始,2015年获批移植者一线从诱导开始,2021年121亿美元专利到期,2024年58亿美元。MM收入占比90%。
达雷妥尤单抗
强生。抗CD38单抗,25年预计140亿美元销售,增长20%,美国占56%。适应症有MM和AL,MM估计有130亿美元的销售。D-Rd 在 MAIA 随访里中位治疗时长≈47.5 个月。2029年专利到期,皮下注射32年到期。
不移植者(D-Rd)第1年(23针):毛额 23.8 万美元;净额(按降20%) 19.0 万/人·年。其后(13针/年):毛额 13.4 万;净额 10.7 万/人·年。
移植者(D-VRd→D+R维持)第1年(16–22针):毛额 16.5–22.7 万;净额区间 13-18.2 万/人·年。第2年起若处于维持(13针/年):毛额 13.4 万;净额区间 10.7 万/人·年。
2012-08-30,Genmab 将达雷妥尤单抗的全球开发与商业化权以独家许可授予强生,首付5500万美元,里程碑10亿美元,分成12-20%。2015年美国上市后的4年时间,上市了8个适应症,2个剂型。
美国首批上市(首适应症):2015-11-16。皮下制剂 Faspro 上市:2020-05-01
美国各适应症/方案的获批时间线(MM)
复发/难治(R/R)
单药 D(≥3线,含PI与IMiD或对二者双耐):2015-11-16(加速批准)。
D+Rd(既往≥1线后):2016-11-21。治疗时长34.3月
D+Vd(既往≥1线后):2016-11-21。治疗时长13.4月
D+Pd(既往≥2线且含来那度胺与PI):2017-06-16。治疗时长11.5月
D+Kd(carfilzomib+dex)(既往1–3线后):2020-08-20。治疗时长11.8月
一线(新诊断,NDMM)
D+VMP(不适合移植,TI):2018-05-08(ALCYONE)。 治疗时长14.7月。
D+Rd(不适合移植,TI;MAIA):2019-06-27。治疗时长47.5月。
D+VTd(适合移植,TE;诱导+巩固,CASSIOPEIA):2019-09-26。
D+VRd(适合移植;诱导+巩固,TE;PERSEUS):2024-07-30。诱导中位时长 6.1 个月,巩固在移植后30–60天启动,再过~2个月进入维持,真实世界71.1%患者在随访期接受了移植,2年OS率:D-VRd 94% vs VRd 91%;4年PFS率:D-VRd 84.3% vs VRd 67.7%。
AL 轻链淀粉样变(Faspro + VCd;加速批准):2021-01-15。治疗时长21.3月
当前一线标准治疗MM
适合移植
首选路径:D-VRd → 采干 → ASCT(Mel200预处理,自体移植)→ D+R维持(MRD指导)。D-VRd(达雷妥尤单抗+硼替佐米+来那度胺+地塞米松)显著提高MRD阴性率并显著延长PFS;方案含D+R维持,并设MRD阴性持续≥1年的停D策略。该组合已进入2025年NCCN首选。
皮下注射版达雷妥尤单抗:1800 mg 皮下注射,C1–2(第一和第二周期) 每周一次,C3–6 每2周一次,从C7起每4周一次;维持期按每4周一次。
硼替佐米(V):1.3 mg/m² 皮下,每周一次(多数中心用D1/8/15;也有D1/4/8/11传统法),每周一次给药神经毒性更低。
来那度胺(R):25 mg,D1–21,Q28d;肾功能减量。
达雷妥尤单抗(d):40 mg 每周;≥75岁多用20 mg。
周期数:诱导4个周期 → 采集造血干细胞 → ASCT(Melphalan 200 mg/m²)→(可加2周期巩固)→ D+R维持
维持
D+R:DARA每4周+来那度胺10–15 mg d1–21;PERSEUS采用MRD指导:若维持12个月仍持续MRD阴性,可考虑停D,只保留R;部分队列R设上限36个周期。
重要备选
Isa-VRd(异沙妥昔单抗+VRd):Ⅲ期 IMROZ 显示对VRd有显著PFS获益,是抗CD38四联的另一选择。
VRd三联:经典基准;SWOG S0777 证实较Rd显著延长PFS/OS;ASCT时机可早可延(DETERMINATION:早期ASCT较延迟ASCT PFS更长,但OS无显著差异)。适合不便用抗CD38者。
高危细胞遗传学优先:可考虑KRd骨架(如FORTE:KRd+ASCT带来更深反应;ATLAS:KRd维持优于单用R,特别在高危亚组)。心脏合并症要谨慎用K。
不适合移植
首选路径1:D-Rd 持续治疗直至进展
证据:MAIA显示,D-Rd较Rd显著延长PFS并提高OS,HR≈0.67。对≥75岁老年组也有一致获益。
剂量(28天/周期):
DARA SC 1800 mg:C1–2每周,C3–6每2周,C7+每4周;
Len 25 mg D1–21(肾功能分层减量);
Dex 40 mg每周(体弱/高龄20 mg)。
首选路径2(身体更好的):Isa-VRd 或 D-VRd
2025年NCCN更新把Isa-VRd列为<80岁且非脆弱患者的Category 1首选。用于“够健壮、想更深缓解”的人群。
对非常高龄/脆弱者的简化路径:VRd-lite 或 渐进式D-Rd
VRd-lite:V 1.3 mg/m² 皮下每周一次;Len 起始10 mg(耐受后升15 mg);Dex 20 mg每周;通常9个周期后转Len维持。
若神经病变明显或极脆弱,D-Rd往往耐受性更好(避开V-PN),但仍需做VTE与感染预防。
小结:不适合移植的人群,脆弱的用D-Rd、好点的用Isa-VRd 或 D-VRd;适合移植的用D-VRd。也就是说大部分一线都会用到抗CD38。
二线路径
目前最常用的一线方案:不移植的一线方案D-Rd(一直用到进展)是2019年MAIA实验确立的,PFS 5年。美国每年新发3.6万人,移植8000人,不移植的有2.8万人。这些人从2025年开始陆续进入D+R耐药,效果更好的Isa-VRd 或 D-VRd是2024年和2025年,5年PFS率有60%+,预计耐药得2030年以后了。移植者,2019年批准D-VTd方案,PFS 7年,2024年D-VRd方案确立,移植者美国批准的维持是用来那度胺单药,但D+R属于2A,也用的逐渐增多。因此,从2025年开始,随着一线方案的逐步进入耐药,因此扩大到每年2.5万人的来那度胺 + 达雷妥尤单抗双耐药是至少的(假设70%最终能走到二线耐药市场)。
因为是四药联用,耐药的规则是:谁在进展发生时仍在用,或停药 ≤60 天,就按谁“耐药”。比如:D-Rd 维持中进展,则来那度胺 + 达雷妥尤单抗双耐药。D-VRd/Isa-VRd 诱导期内就生化上升/进展,三类同耐药(来那度胺+硼替佐米+达雷妥尤单抗双耐药)。完成诱导→ASCT 后,仅来那度胺维持,CD38/PI早停,现复发,来那度胺 耐药。因中性粒细胞低停了 CD38,仅 Rd 继续,随后进展,来那度胺耐药。不耐受归因:看毒性,按减量、间歇给药等。
来那度胺和达雷妥尤单抗都耐药的治疗,这个人数将逐步成为最多,因为一线维持疗法以这俩药为主。美国 NCCN Preferred(首选)是CAR-T 、双抗和 无 CD38、且不含 Len 的三联。
CAR-T
2024-04-05 NCCN把两款 CAR-T放到更早治疗线,列为 Category 1,放进Preferred(首选)在≥1 线的耐药市场,意味着CAR-T进入二线市场。CARTITUDE-4 随机Ⅲ期已证明 cilta-cel 显著优于标准三联(如 PVd/DPd)在 PFS/OS 上,这也是许多中心把CAR-T优先的现实依据。靶点是 BCMA,在浆细胞/骨髓瘤细胞上90%表达。cilta-cel 在主要注册/确证研究中的患者年龄中位数基本都在 61 岁左右,真实世界64-65岁。
目前CARVYKT和ABECMA的药品单人净销售在40万美元左右(目前一线的D+R的维持期间,一年净销售额在23万美元,明年来那度胺仿制放量后估计更低)。治疗的门诊费用在3-5万美元,住院10-15万美元,人次,门诊与住院各半。在美国可以报销,有医保的个人支付最多不超过1万美元,但医保需事前授权。目前CAR-T实施的问题:回输后至少2周留在医疗机构附近,且前7–10天每日门诊评估,看护人 24/7 在侧。已取消 REMS(2025-06),但实际仍由厂家认证治疗中心或具备相应能力的医院实施。制造与排期耗时,可能需要1-2月,真实世界可能疾病进展等不及。“T 细胞恶性肿瘤”黑框警示。双抗的分流。不是所有人的体力都能抗住,3天淋巴清除化疗+可能危及生命的急性毒性+连续密集监测与后勤要求;体能差/器官功能边缘的患者更容易在等待或回输后并发症失代偿/无法完成疗程。放行中位44天、采集至回输79天,治疗前需桥接1个周期。
Cilta-cel(Carvykti)
2024-04-05:FDA 扩大适应证至“≥1 线且 Len-R(既往含 IMiD+PI),不需要额外满足“CD38暴露/耐药”的前提。2024-09(NCCN v4.2024):把 cilta-cel 列为 Category 1 的“既往治疗后、Len-R(≥1 线)”选项。CAR-T 放进“Preferred(首选)”分组。
2023年披露核心 III 期:CARTITUDE-4,cilta-cel(一次性输注;允许短程过渡治疗) vs 医师预先选择的 DPd 或 PVd,61 岁,既往1/2/3 线:约 33%/40%/27%。既往 抗-CD38 暴露 ~25%;三类药暴露 ~26%;Len-R 100%。30个月PFS率:59.4%(cilta-cel)vs 25.7%(对照)。30个月OS率 76.4% vs 63.8%。ORR / CR或更好:84.6% / 76.9%(cilta-cel) vs 67.3% / 24.2%(SOC)。30个月DOR率 67.4% vs 35.5%。MRD 阴性,10^-6 灵敏度:85.6% vs 18.6%。安全性:CRS 常见、以 1–2 级 为主;任何级别 ~75–80%,≥3 级 ~1–3%;SPM(二次原发肿瘤):中位随访 33.6 个月 时,cilta-cel 组 13.0%、对照 11.5%(其中血液系统 SPM 3.4% vs 0.5%)。
在之前的二期CARTITUDE-1,97 例重度 RRMM(中位既往 6 线;三类药难治 88%;五药难治 42%)。疗效(28–33 个月):ORR ~98%;sCR ~80–83%;mPFS 34.9 个月。5 年随访(EHA 2025),中位 OS 60.7 个月(≈5 年);33% 患者 ≥5 年“无治疗且无疾病进展”;一中心 12 例持续 MRD-阴性+ 影像阴性。
还在开的Ⅲ期:CARTITUDE-5(MMY3004;NCT04923893),2021年开始的初治、无移植计划(NTE-NDMM) 方案:VRd 诱导 → cilta-cel vs VRd 诱导 → Rd 维持;主要终点 PFS;全球、多中心、开放随机;入组743,2024.7完成入组,随访中。VRd 组在以前的CEPHEUS临床中做过PFS 52.6 月。无法判断能否一定阳性。目前不移植的一线方案是D-Rd PFS 61.9月,后面DVRd 54月 PFS 率 68.1%。
还在开的Ⅲ期:EMAGINE / CARTITUDE-6(MMY3005/EMN28;NCT05257083),初治、可移植(TE-NDMM) 方案:DVRd → cilta-cel vs DVRd → 自体移植(ASCT);主要终点 PFS;全球、多中心、开放随机,2023-10 开始入组,已完成入组,由欧洲骨髓瘤网络 EMN作为赞助方/主导,强生提供药物和资助。无法判断PFS 能否跑赢。4年PFS率:D-VRd 84.3%。
anito-cel(CART-ddBCMA)
Arcellx 与 Gilead 旗下 Kite Pharma 共同开发
三期:iMMagine-3,复发/难治MM,既往1–3线,且既往暴露过IMiD和抗CD38,2024-10-31 已开始入组。早期临床数据:ORR 95–97%,CR/sCR 62%;6个月PFS率≈90%、OS率≈95%,MRD阴性 92%,安全性:任意级CRS 84%(大多0–1级),ICANS 9%(3级≈2%);到披露时未见延迟型神经事件。
安全性方面,Cilta-cel与anito-cel的对比:
CRS:主要是发烧和低血氧。Cilta-cel的综合 CARTITUDE-1&4:CRS 84%,≥G3 4%;中位起始 7 天(1–23),中位持续 4 天。CRS发作时间7天使得可以在门诊输注,不用立即住院。anito-cel:CRS 94%(100M 剂量组),多数为 G1–2;RP2D 未见 ≥G3 CRS;中位起始 2 天,持续约 5–6 天。注:Ⅱ期早期摘要曾报告 3 例死亡,其中 1 例归因 CRS。
ICANS:神经毒性综合征。Cilta-cel:ICANS 13%,≥G3 2%;中位起始 8 天(1–28),中位缓解 3 天。anito-cel:Ⅰ期:ICANS 16% G1–2、3% G3(100M 组);RP2D 报告仅 1 例 G3 ICANS,起始约 4–7 天。
帕金森样综合征:cilta-cel:3%(8/285),≥G3 2%(5/285);中位起始 56 天;已报道的病例中恢复率很低(1/8)。CARTITUDE-4 的“运动/认知类MNT(含帕金森样)”发生率显著低于 CARTITUDE-1(约 0.6% vs 6%),桥接降负荷、早期干预CRS/ICANS、强化监测。颅神经麻痹—cilta-cel:合并CARTITUDE-1+4披露为7%(19/285),≥G3 1%,中位起始约21天、中位缓解66天。anito-cel目前未见:Ⅰ/Ⅱ期披露中未观察到帕金森样、颅神经麻痹或 GBS 等“迟发非 ICANS 神经毒性”。anito-cel的iMMagine-1明确把任何“现有或既往的中枢神经系统(CNS)病变”都排除。iMMagine-3三期临床正在做,没有排除。
ide-cel(Abecma)
Bristol Myers Squibb(BMS,含其子公司 Celgene)与 2seventy bio(bluebird oncology 分拆)合作,疗效不如Cilta-cel。用于经≥2 线治疗后且既往暴露过 IMiD+PI+抗CD38 的 R/R MM 成人。KarMMa-3(Ⅲ期,2–4L TCE vs 医师选择 SOC,NEJM 2023):中位 PFS 13.3 vs 4.4 月,HR 0.49;ORR 71% vs 42%,≥CR 39% vs 5%。早期随访曾见OS 曲线前期不利(早期死亡事件更多)。KarMMa(Ⅱ期,4L+ TCE,NEJM 2021):ORR 73%,≥CR 33%,MRD-阴性 26%(按全部给药人群),中位 PFS 8.8 个月;后续更新 中位 OS 24.8 个月。
小结:来那度胺和达雷妥尤单抗都耐药,目前效果最好的方案是Cilta-cel,但CAR-T的可及性使得其渗透率是有天花板的。强生和传奇联合开发。今年估计得有5000人治疗吧(美国收入占84%,按海外价比美国低30%估计,美国有4000人),2024年美国估计也就2000例吧,美国的渗透确实很快。Cilta-cel未来一线能否赢,不会判断。anito-cel的安全性似乎要好于Cilta-cel,但进度整体起码要慢4年
双/三特异性抗体
患者不适合/无法获得CAR-T(器官功能/照护条件/等候期/产能)、或需现成起效。
Linvoseltamab:BCMA×CD3 TCE,再生元
2025-07-02 被 FDA 以“加速批准”核准用于既往≥4线且包含 PI、IMiD、抗CD38 的复发/难治MM成人患者。被NCCN新增为“Preferred”(在≥4线后人群)。
Ⅱ期纳入:RRMM 且≥3 线进展或 TCR(三类耐药);200 mg 队列 n=117,中位年龄 70 岁,高危细胞遗传 39%,penta-refractory 28%。200 mg 队列 n=117,中位既往 5 线,TCR 80%,高危细胞遗传 39%,penta-refractory 28%。≥三类难治 82.1%。
ORR 71%;≥CR 50%(中位随访 14.3 月)。ORR 71%;≥CR 50%。 DOR 中位 29.4 月(在取得反应者,n=83)。12 个月 PFS 率 70%。OS:中位 31.4 个月;12 个月 OS 率 75.3%。
CRS:46.2%(G1 35.0%、G2 10.3%、G3 0.9%),以低级别为主。ICANS:7.7%(G1/2/3 各 2.6%)。感染:任何级别 74.4%;≥G3 33.3%(G4 2.6%);严重感染的发生率与严重度随时间下降。 血液学毒性:中性粒细胞减少 G3 18.8% / G4 23.1%;贫血 G3 30.8%。阶梯预处理给药:Day1 5 mg → Day8 25 mg → Day15 首个 200 mg;随后200 mg QW(第4–13周,共10次)→ Q2W;达到且维持 ≥VGPR(第24周起且累计≥17次 200 mg)可改为 Q4W。前两次阶梯剂量各需住院观察 24 小时。
EMN39 / NCT06932562(Ⅲ期,随机、开放),设计:DRd(达雷妥尤单抗+来那度胺+地塞米松)诱导后转入 linvoseltamab vs 继续DRd,人群为新诊断且不适合自体移植(TI-NDMM);目标样本量约1000,主要终点为疗效(以PFS为主)。状态:Phase 3,Active/启动中。
Teclistamab / Talquetamab:强生,BCMA×CD3和GPRC5D×CD3(在MM 90%+表达)已上市
NCCN将其列为“useful in certain circumstances在特定情况下可用”用于≥3线后重度预治人群。位置低于“Preferred(首选)”与“Other recommended(其他推荐)。在 NCCN 被列为 Preferred,Category 2A。Tecvayli:标签提示严重/致命感染风险(≥G3感染 35%、致命 4.2%),并要求阶梯起始且每次阶梯剂量后住院监测48小时。Talvey:同样阶梯起始需48小时住院;且有口腔/味觉异常、皮肤/甲改变与体重下降等GPRC5D特异毒性(如任一口腔毒性约 80%、体重下降 ~62%)。这些因素限制了广泛一线之选的可行性。
Teclistamab(Tecvayli),BCMA×CD3,NEJM 报道(扩展集N=165,中位随访14.1月):ORR 63.0%;≥CR 39.4%;mDOR 18.4月;mPFS 11.3月;MRD阴性 26.7%。不良反应:CRS 72.1%(≥G3 0.6%)、ICANS 3.0%(均1–2级)、感染 76.4%(≥G3 44.8%)。既往治疗线数:中位 5,三类药暴露 100%;三类药难治 77.6%,抗CD38 单抗难治 89.7%,高危细胞遗传 25.7%;骨髓外浆细胞瘤 17%,既往自体移植 81.8%,对上一线难治 89.7%。
MajesTEC-7(NCT05552222;Ⅲ期,NDMM、不可移植或不打算移植)设计:Tec-DR(teclistamab+daratumumab+来那度胺)、以及Tal-D(talquetamab+daratumumab+来那度胺),与 DRd 对比;主要终点为疗效(PFS 等),已完成安全性“run-in”并公布可管理的早期数据。
MajesTEC-4 / EMN30(NCT05243797;Ⅲ期,移植后维持)设计:teclistamab+来那度胺、teclistamab 单药 vs 来那度胺单药 作为ASCT 后维持;主要终点 PFS;已公布安全性 run-in 为“可行/可管理”。
Talquetamab(Talvey),GPRC5D×CD3双抗,Q2W:ORR 71%;≥CR 43%;mDOR 17.9月,QW:ORR 73%;≥CR 35%;mDOR 10.2月。24月OS率67%。安全性:(N=339,总体):CRS 76%(≥G3 1.5%);味觉障碍 70%、甲/指甲异常 50%、皮肤异常 41%、皮疹 38%、体重下降 35%、口干 34%;感染(细菌含败血症)19%(≥G3 9%)。既往治疗线数:中位 5(4–13),三类药难治约 73%,高危细胞遗传 29%,既往自体移植 78%,对上一线难治 94%。
Elranatamab:BCMA×CD3,已上市,辉瑞
FDA 加速批准:≥4 线 RRMM,且包含PI + IMiD + 抗CD38 单抗,Cohort A(BCMA-naive,n=123;有效性人群 n=97,ORR 61.0%;≥CR 35–37%(随访延长后 37.4%)。 中位随访延长至 28.4 个月时:中位PFS 17.2个月;中位OS 24.6个月;24个月持续缓解率 66.9%。安全性(任何级别/3–4级):感染 70.7%/42.3%(G5 6.5%);CRS 57.7%(均≤2级);ICANS 4.9%(均≤2级);中性粒细胞减少 49.6%/49.6%。转为Q2W后3–4级AE比例下降。中位 5 线(2–22);三类药暴露 100%。
Cohort B(既往BCMA治疗,n=63 计入分析),ORR 33.3%;9个月缓解维持率 84.3%;人群中既往接受过 BCMA-ADC 73%、BCMA-CAR-T 32%。 标签整合安全性(n=183):CRS 58%(3–4级0.5%)、疲劳 43%(3–4级6%)、注射部位反应 37%、腹泻 36%;实验室学 3–4级:淋巴细胞减少 84%、中性粒细胞减少 51% 等。中位 8 线(4–19)。
列入“晚期复发(>3线)”部分的“Preferred(首选)”且Category 2A(一致同意、证据等级较低);同时强调适用人群需满足已接受 ≥4 线且包含 CD38 mAb、PI、IMiD这一获批人群。
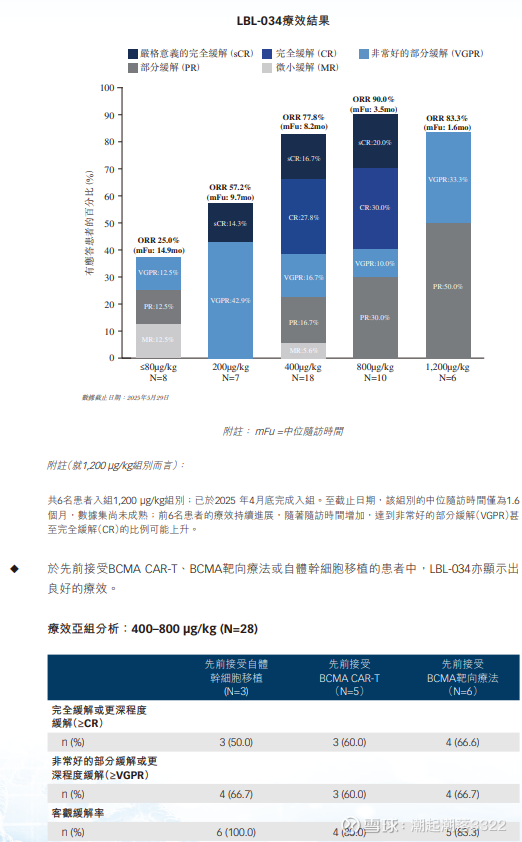
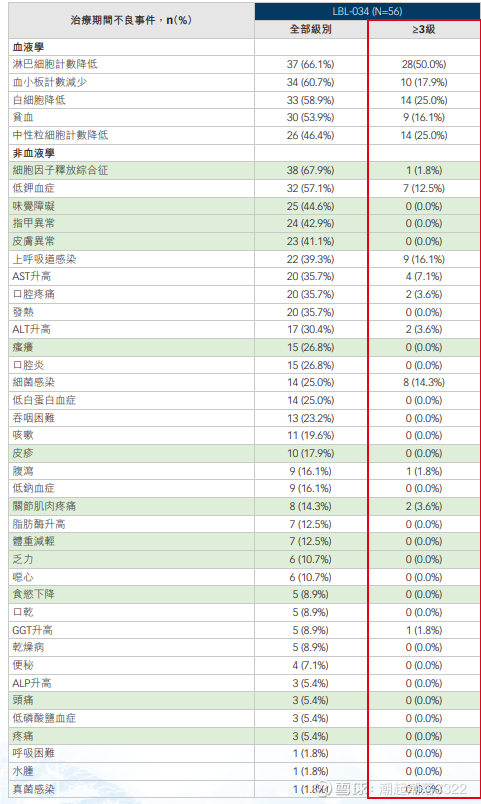
LBL-034,不对称结构 的 GPRC5D×CD3,维立志博
截至2025年5月29日,400-800ug的ORR是82.1%,400ug(18例)的orr是78%,≥VGPR61%,≥CR 44%。800ug(n=10)的ORR是90%,≥VGPR 60%,≥CR 50%。1200ug未观察到限制性毒性,1200ug入的是髓外瘤,最差的情况。强生的双抗只做到400-800ug。将CD3的亲和力降低到-8次方,从而降低副作用。8月开始入组二期临床。后面做BCMA经治和髓外瘤(10–15%)。CRS三级1.8%。疗效和安全性不错,等待更多数据披露。
ABBV-383:BCMA×CD3,双价高亲和BCMA臂 + 低亲和CD3臂 + 静默Fc,公司:AbbVie
入组 RRMM、既往≥3线且PI/IMiD/抗CD38均已用,未暴露BCMA疗法。基线中位既往5线,80% 三类药难治。RP2D 60 mg Q4W 时给出:ORR ≈65%、≥VGPR ≈50%。60 mg:中位 PFS 11.2 月;12 月 DOR 66%。CRS:50–71%(大多 1–2级;各剂量层未见 4/5 级);ICANS 3–5%(个别 60 mg 层现 3 级,无 4/5 级)。感染:总体 56–71%,≥3级 25–34%
Ⅲ期临床(进行中)CERVINO;NCT06158841:etentamig 单药(Q4W)vs 医师选择可用标准方案,3L+ RRMM,随机、开放、多中心,目标样本量约 380 例;2024-06-05 AbbVie 宣布已启动并入组。定位优势:月给药(Q4W)、起初不需步进给药
JNJ-79635322,BCMA×GPRC5D×CD3,强生
既往4 线,三类暴露(PI/IMiD/抗CD38)100%,67岁。皮下 100 mg Q4W,仅需一次 5 mg 阶梯剂量,在BCMA/GPRC5D 未暴露且按 RP2D(100 mg Q4W)给药的队列(n=27):ORR 100%;≥CR 70.4%;≥VGPR 96.3%。12 个月 PFS 95.0%(所有剂量汇总 74.1%)。在既往用过 BCMA/GPRC5D的患者中(50–300 mg 汇总):ORR 55.0%。CRS:总体以 1–2 级为主;在未用预防托珠单抗的 100 mg 组,CRS 69%(多为 1 级;无 ≥3 级);加用预防托珠单抗后降至 20%(均为 1 级)。ICANS:RP2D 组未观察到 ICANS。RP2D 组任意级 80.6%,感染:≥3/4 级 33.3%;曾有1 例肺炎致死(VGPR 中发生)。低丙球常见(RP2D 组 50%),约 47% 用过 IVIG 支持。口腔/味觉相关不良反应(GPRC5D 相关):有发生,但频率与严重度低于 talquetamab;体重下降多为 1–2 级、比例较低。
BCMA 靶向 ADC
GSK的Belantamab,联合方案(BVd、BPd)写入美国标签,用于≥1 线复发/难治 MM。此申请正由 FDA 审评。目前FDA在犹豫要不要给批准,DREAMM-7/8 显示 显著 PFS 获益,且 DREAMM-7 有 OS 优势,但是眼部毒性发生率高、复发多、部分未完全恢复;剂量/给药间隔探索不足,现提报剂量的可耐受性较差,需要更合理的剂量优化。英国、欧盟、日本已经上市。
DREAMM-3(Ⅲ期,单药 vs Pd),≥2 线复发难治(RRMM),PFS:未达优效;HR 1.03。发现不联合Pd和Vd,疗效就发布不出来。后面V药也踩过这个坑。
DREAMM-7(BVd vs DVd):≥1 线 RRMM,mPFS 36.6 vs 13.4 月;HR 0.41,18 月 OS 率 84% vs 73%。安全性:眼部事件更常见于 BVd(79% vs 29%),总体经剂量调整可管理。≥3 级 AE 95% vs 78%。
DREAMM-8(BPd vs PVd):既往≥1 线且用过 Len 的 RRMM。主要终点(PFS):HR 0.52(0.37–0.73;P<0.001);12 月 PFS 率 71% vs 51%。
DREAMM-10(BRd vs DRd,TI-NDMM,一线):正在Ⅲ期招募;主要看 PFS/MRD-阴性
无 CD38、且不含 Len 的三联
KPd(卡非佐米+泊马度胺+地塞米松):NCCN,Preferred(首选),适用于“CD38 难治”列;同时也列入Len 难治”列的 Preferred。属 Category 2A。已治疗 1–3 线的早期复发人群。没有独立的 FDA 批准适应症,属于超说明书用法。没有做三期临床。
KPd的SELECT 研究(Ⅱ期,多中心):既往暴露:来那度胺 100%,硼替佐米 87%,达雷妥尤单抗 77%,卡非佐米 4%,37% 为三重耐药(来那度胺 ,硼替佐米 ,达雷妥尤单抗);难治:来那度胺难治 100%, 达雷妥尤单抗难治 75%(40 人暴露中 39 人难治),硼替佐米难治 35%,卡非佐米难治 2%。中位 68 岁,1 线 17%,2 线 83%。ORR 58%(≥VGPR 35%,≥CR 6%,ORR未达预设),mPFS 11.1 月,mOS 18.8 月;MRD<10⁻⁵ 阴性 10%。≥3级TEAE 67%。Amgen,卡非佐米是选择性蛋白酶体抑制剂,没有开三期。比起硼替佐米,周围神经病变:≥2级 PN:6% vs 32%,输液。
Kd(卡非佐米+地塞米松)列入Other recommended(其他推荐),但证据级别为 Category 1。已治疗 1–3 线;作为len/CD38 均回避时的非IMiD 双联备选。适应症:用于已接受 1–3 线治疗的复发/难治多发性骨髓瘤(RRMM)成人患者。Kd(n=464)对照 Vd(n=465),既往来那度胺/硼替佐米38% / 54%。PFS(主要终点):18.7 vs 9.4 个月,OS:47.6 vs 40.0 个月,这个临床达雷妥尤单抗无暴露,代表性不足。
PVd(泊马度胺+硼替佐米+地塞米松):NCCN,在Len 难治列为 Preferred,Category 1(基于 OPTIMISMM Ⅲ期证据);在CD38 难治”列**同样列为 Preferred,Category 2A。已治疗 1–3 线;对既往用过 len 的复发/len-难治尤为适配。没有独立的 FDA 批准适应症,属超说明书联合。
PVd的OPTIMISMM三期临床:2018年披露,PVd vs Vd,既往治疗线数中位 2,1 线:40% vs 41%;2 线:42% vs 37%;≥3 线:19% vs 21%,67 vs 68 岁,全部患者既往来那度胺暴露,且来那度胺耐药约 70%,达雷妥尤单抗耐药比例=0%,PFS:11.20 vs 7.10 月,来那度胺耐药:9.53 vs 5.59 月。这个临床达雷妥尤单抗无暴露,代表性不足。
注:PVd是在Len 难治和CD38 难治都是Preferred(优先选),但Len 难治的证据级别是1(最高),CD38是2A(证据较低但一致)。由于fda没有获批,属于超适应症,医生可以开,企业不能宣传。Category 1/2A 的 off-label 往往可获报销(仍可能要事前授权、按计划条款走流程)。
EPd(艾洛珠单抗+泊马度胺+地塞米松):推荐级别:在CD38 难治与Len 难治两列均列入 Preferred(多为 Category 2A)。场景:已治疗 1–3 线;尤其CD38-单抗难治后作为非CD38 免疫通路的 IMiD 组合。
EPd(艾洛珠单抗+泊马度胺+Dex,ELOQUENT-3):67 岁,既往治疗线数中位 3。来那度胺难治 87%;PI 难治 80%;双重难治(Len+PI)70%。既往 SCT 55%,用过硼替佐米 100%、来那度胺 99%、卡非佐米 21%、达雷妥尤单抗 3%。PFS:10.3 vs 4.7 个月,ORR:53.3% vs 26.3%,OS:29.8 vs 17.4 个月,2018年已获 FDA 批准,适用于 至少接受过两线治疗,且既往用过来那度胺与 PI 的成人 RRMM。艾洛珠单抗是BMS的,人源化 IgG1 单克隆抗体,靶点为 SLAMF7,输液。这个临床达雷妥尤单抗暴露太少,代表性不足。
IPd(伊沙佐米+泊马度胺+地塞米松)在CD38 难治与Len 难治两列均为 Preferred、Category 2A已治疗 1–3 线;全口服三联,适合门诊便利性需求且回避 len/CD38 的人群。没做三期,没有 FDA 批准适应症
二期临床Alliance A061202,设计/人群:Len难治、第一复发RRMM,IPd vs Pd,ORR:63.2%(IPd)vs 43.6%(Pd)。≥VGPR:28.9% vs 5.1%(P=0.0063)。中位PFS:20.3 vs 7.5个月;HR 0.437血液学不良事件↑(2–4级):中性粒细胞减少 71.1% vs 46.2%、淋巴细胞减少 63.2% vs 35.9%、贫血 34.2% vs 20.5%、血小板减少 13.2% vs 5.1%。这个临床达雷妥尤单抗无暴露,代表性不足。
Selinexor 组合(如 XVd=Selinexor+Vd):现归入 Other recommended,但证据级别仍为 Category 1(BOSTON Ⅲ期一次/周给药方案)线别/场景:已治疗 1–3 线;len-spar ing、CD38-free 的一类口服+PI组合备选。
XVd(塞利尼索+硼替佐米+地塞米松):2022年披露,BOSTON Ⅲ期随机对照Vd(硼替佐米+地塞米松),年龄中位 66–67 岁。既往治疗线数:1 线 51% vs 48%;2 线 33% vs 31%;3 线 16% vs 21%。既往暴露:硼替佐米 68.7% vs 70.0%;来那度胺 39.5% vs 37.2%;达雷妥尤单抗 5.6% vs 2.9%。ORR(IRC):76.4% vs 62.3%,PFS(主要终点):13.93 个月 vs 9.46 个月,HR 0.70。DoR(应答持续时间,中位):20.3 个月 vs 12.9 个月,FDA 扩展适应症批准(SVd,≥1线后)于2020-12-18。塞利尼索抑制XPO1/CRM1,开发公司是Karyopharm。这个临床达雷妥尤单抗暴露太少,代表性不足。
可见以上的三联方案大部分没有做 达雷妥尤单抗暴露的临床。
BCL-2抑制剂
亚盛2575在2024年ASH会议披露MM的数据
2024-11-05 数据截止,n=58,既往治疗线数中位为 3;MM 48,AL 10。Arm A(Lisaftoclax + Pd):41 例;剂量分布 400 mg×3 / 600 mg×4 / 800 mg×15 / 1000 mg×13 / 1200 mg×6;可评 36 例。Arm B(Lisaftoclax + DRd):7 例(均 600 mg);可评 5 例。有效性(Arm A,RRMM):ORR 63.9%(23/36);≥VGPR 30.6%(CR 3,VGPR 8;PR 12)。中位 PFS 9.7 个月。32 名用抗 CD38 单克隆抗体预处理的 RRMM 患者的反应率,发现 ORR 为 62.5%。该组的中位 PFS 为 9.7 个月。中位随访时间为 9.2 个月,A 组在 400 mg、600 mg、800 mg、1000 mg、1200 mg 和总剂量组分别为 6.5 个月、7.4 个月、11.1 个月、7.0 个月和 9.7 个月。有效性(Arm B,RRMM):可评 5 例中 CR 40%。接受利沙托克加 Pd 的 AL 淀粉样变性患者的 ORR 为 400 mg 为 0% (n = 1),600 mg 为 100% (n = 4),800 mg 为 100% (n = 2),1000 mg 为 100% (n = 2),全组 (n = 9) 的 ORR 为 88.9%。总体缓解时间中位为 0.9 个月。治疗相关不良事件 67.2%;≥3 级 24.1%;其中≥3 级中性粒细胞减少 13.8%、发热性中性粒细胞减少 1.7%;DRd 臂出现 1 例 DLT:QTc 延长;未见明显 DDI 信号。Ailawadhi 指出,“对患者来说,第一大诱惑因素是全口服方案”,并且该试验的入组人数非常活跃。基线:A组的所有患者既往接受过蛋白酶体抑制剂治疗,95.1%的患者既往接受过免疫调节剂治疗,85.4%的患者既往接受过抗CD38抗体治疗。只有 4 名患者患有 t(11;14),其中3人PR或更好,1人病情稳定。基线:n=52,中位年龄 69.5 岁;既往治疗线数中位 3,2024-05-29。网页链接
维纳克拉的2个失败的MM临床:
BELLINI(NCT02755597),RRMM,既往 1–3 线;t(11;14) 占比(基线):约 11%–16%。对照 Placebo+Vd(硼替佐米+Dex)。样本量 n=291(Ven 194 / Pbo 97)。Ven 剂量 800 mg QD;Vd 21 天 ×8 周期后转 35 天周期。主要终点:PFS。PFS:23.4 vs 11.4 月;HR 0.58,OS:HR 1.19,p=0.39。治疗相关早期死亡在 Ven 组集中(TE 死亡 7% vs 2%),但随时间推移 OS HR 改善至 1.19(仍未优于对照)。用800mg每天的剂量,结果毒性太大,导致OS没有优势。2019 年中期分析显示 感染相关死亡增加,FDA发出安全警示并暂停入组。PFS 23月是因为基线:既往治疗线数:1 线约 47%(对照 45%);2–3 线 53%(对照 55%)
t(11;14)亚组: PFS ≈36.8 vs 9.3 月;HR 0.12;OS 趋势 HR 0.61(0.16–2.32)。BCL2 高表达:98人/291,PFS 22.4 vs 9.9 月;HR 0.24。非 t(11;14) 且 BCL2 低:PFS 15.3 vs 12.2 月;HR 0.81
≥3 级血液学:中性粒细胞减少 30% vs 8%;血小板减少 26% vs 40%。致死 AE(治疗相关):Ven 2%(肺炎×2、MODS+脓毒性休克×1、未指明×1) vs 对照 0%。≥3 级感染:肺炎 16% vs 9%。≥3 级腹泻 15% vs 11%。
CANOVA(NCT03539744),Venetoclax 800 mg QD + DEX(VenDex) vs 泊马度胺 4 mg d1–21 Q28 + DEX;均为t(11;14)+ RRMM,既往≥2 线,均经PI暴露、对Len复发/难治。样本量 n=263。mPFS(月):9.9 vs 5.8(HR 0.82);BCL2-high:9.4 vs 3.8(HR 0.64)mOS(月):32.4 vs 24.5(HR 0.70),ORR(≥VGPR):62%(39%) vs 35%(14%);BCL2-high:69%(48%) vs 36%(13%),VenDex 的治疗暴露时长更长(9.1 vs 4.3 月),但≥3级不良事件总体更少:67% vs 83%;然而治疗相关死亡在 VenDex 更高:8%(11例) vs 3%(4例),其中肺炎相关占多数。终点是PFS,未达显著。主要是临床设计中,治疗组没有加泊马度胺,对照组挺强。
NCCN里,V药在MM的定位是——仅限 t(11;14)+ 的既往治疗后复发/难治患者,列为在特定情形可用,等级为 Category 2A;形式为 V药 + Dex,± D 或 蛋白酶体抑制剂(如硼替佐米/卡非佐米)。不是“Preferred”,且在MM领域仍属非注册标签外使用。
总结:
1.从MM的发展历史看,2000年左右,MM治疗的历史就进入诱导+移植或者不移植+维持的阶段。方案从沙利度胺+地塞米松->硼替佐米+沙利度胺+地塞米松->硼替佐米+来那度胺+地塞米松->达雷妥尤单抗+硼替佐米+来那度胺+地塞米松。这之后,靶向BCMA的CAR-T和TCE双抗,不断把治疗线前移,正在进行挑战一线的三期临床。OS从90年的年代的3年提升到移植者5年OS率80%,不移植者D-Rd方案的 中位OS 90月。这个过程诞生了2个百亿美元的大药达雷妥尤单抗和来那度胺,来那度胺凭借更好的疗效和更低的神经毒性替代了沙利度胺的位置,达雷妥尤单抗带来新的抗CD38的机制对疗效进行加成而毒性可耐受,进而发展成了四联。但目前来那度胺21年专利到期,达雷妥尤单抗29年专利到期,皮下注射32年到期,泊马度胺专利到期。在CAR-T领域,强生和吉利德分别拿下Cilta-cel与anito-cel各一半的权益,TCE双抗和ADC的竞争就更激烈了,强生、吉利德、再生元、辉瑞、艾伯维、GSK都有落子,其中强生的三抗在早期临床的4线人群里做出好数据,ORR100%,1年PFS 95.0%。本质上这些疗法的崛起是靶向BCMA、GPRC5D 结合当下免疫疗法、CAR-T疗法、ADC疗法。
2.不移植者的一线治疗的维持用D+R,估计有2.8万人,移植者8000人的维持治疗的,真实世界有一半是用来那度胺维持,一半用D+R,也就是4000人左右。也就是说一线用D+R维持的新发有3.2万人,按70%耐药后进入二线,人群有2.2万人,t(11;14)染色体结构变异占15-20%,4000-5000人。
3.目前的格局是一线是D-Rd或D-VRd占据,未来二线主要是D+R耐药,TCE双抗、CAR-T、ADC在二线耐药的PFS上有明显的优势,Carvykti的PFS 3-4年,但CAR-T有人群限制,TCE双抗Linvoseltamab(再生元)中位5线,≥三类难治 82.1%,12 个月 PFS 率 70%,但TCE双抗普遍毒性不小,需阶梯给药住院,感染负担重等。BCMA 靶点 ADC也是效果好,但眼毒性大。目前CAR-T、TCE双抗和ADC还在做一线的三期临床,如果真把一线做下来,那么一线的D+R市场会小一些,连带二线耐药市场也会变小。原来早期用于RR MM的三联方案KPd、Kd、PVd、EPd、IPd、XVd,虽然有几个指南Preferred、Category 1推荐,但有的没有FDA获批。Kd 和 XVd,属于FDA二线获批,但之前的三期临床数据的基线主要是30%多的既往R暴露,数据没法对比,NCCN这两个属于Other recommended级别。亚盛2575的思路其实是跟上面的三联或二联方案联用,替代原来三联的RR MM的市场。
4.也就是说2575如果成了,在一部分的二线市场还是能吃到,但空间小了很多,目前不知道2575在针对t(11;14)能做出多少的PFS,能否在这个人群上多加点分。亚盛2575虽然在二线被CAR-T、TCE和部分三联方案挤占空间。但由于属于新机制bcl-2,如果二线被挤占了,可以往三线做,但未来三线的耐药可能就多了一个BCMA的耐药,维立志博已经在考虑做这个耐药了,亚盛毕竟还没开始三期临床,后面假设做出来上市了得4年后了吧,那个时候估计BCMA靶点的药就多了,所以亚盛这个三期临床除了入DRV这些耐药的人群,也可以考虑入点BCMA靶点耐药的,虽然现在这个人群还很少。
5.从亚盛2575目前披露的临床数据看,全口服方案(比较吸引人),2575+泊马度胺+地塞米松,36例,针对D+R 暴露的中位3线人群,400mg-1200mg的PFS是9.7月,预计最后剂量定在800-1000mg,可能PFS还能再往上走走。≥3 级TRAE 24.1%,安全性好。只入组4 名t(11;14),目标是全人群。
6.V药在MM上开过两次三期,都失败了。第一次是其800mg每天的剂量在全人群中因为感染死亡的增加导致OS失败。第二次在临床设计上V药+地塞米松对照泊马度胺+地塞米松,泊马度胺属于三代免疫调节剂,在二线治疗中,就是要替代来那度胺二代药耐药的位置,本身比较强,V药的治疗组又没加上泊马度胺(不知道是否考虑安全问题),最后PFS在t(11;14)人群上没做出差异化。
7.后续2575如果开全人群的D+R耐药的注册临床,做的是二线的话,如果治疗组是2575+泊马度胺+地塞米松(泊马度胺专利已到期,这个临床可能在药费的钱并不多),对照组会选哪个。一般来说泊马度胺+地塞米松是二线/三线公认的有效对照,被多项Ⅲ期注册试验证实并用于对照臂(如 APOLLO:D-Pd vs Pd;ICARIA:Isa-Pd vs Pd),这个对照组弱,之前在仅暴露Len与PI的人群里也就6个多月PFS。还有上文提到的三联方案KPd、Kd、PVd、EPd、IPd、XVd,用于D+R耐药。目前只有 Kd 和 XVd,属于FDA二线获批,之前数据没有做D暴露的。亚盛这个临床的不确定性是OS 不知道会不会受后线影响,毕竟CAR-T和TCE双抗都起来了。
8.简单拍下亚盛2575在MM的市场空间,假设2575用800mg的剂量,美国每mg的上市价格跟V药一样,800mg对应2.4万/月,假设PFS最终能做10个月,关于PFS最后到多少,看上市后有多少比例选择二线,多少比例选择三线及以上,如果是二线的人就用,肯定不止10个月,20个月都有可能,因为10个月的数据的基线是既往治疗线数3,且有低剂量的治疗在,且入组的t(11;14)人群很少。如果后面三线的人群做的多,那么PFS可能就10个月左右了。先按平均12月,对应29万/人年,二线+三线及以后共5000人吧,毕竟是新机制的药,即使二线人少,再后线也能用用,峰值15亿美元的美国市场的概率还是挺大的。