脑机接口、神经疾病的基础知识

一年前开始重点关注脑机接口,意念控制,这是多么科幻、美好的前景。听了很多专家的访谈,一直想写篇这方面的文章。
此前一直想深入分析下$丽珠集团(SZ000513)$ 近期上市/在研的各类药物机理,陆续写了抑郁症—丽珠加油 ,流感疫苗现状及研发 ,丽珠JP1366情况 等篇文章。近年来,丽珠在精神领域着重布局,包括KCNQ 2/3 、阿立哌唑微球、布瑞哌唑微球等,一直想深度科普下这些产品的情况。
但无论是脑机接口,还是神经疾病,都涉及到大脑神经元。这是分析研究的基础,只有弄清楚神经元的机制,才能去理解脑机接口、神经疾病。于是开始查阅这方面资料,边查边理解,写写停停,折腾了几个月,属于写作耗时最长、查阅资料最多的文章之一,形成本文。
本文可能会比较晦涩,希望诸位有耐心看下去。创作不易,欢迎关注、点赞收藏。
一、神经元细胞的理论知识
大脑的主要构成细胞包括神经元细胞、神经胶质细胞、少量血管相关辅助细胞等。神经元是大脑的功能核心细胞,负责信息的接收、传导与整合,是意识和决策的决策者和信号处理器。成年人脑内神经元总数约860亿个,占脑细胞总数的10%-20%。胶质细胞占脑细胞总数的80%-90%,是保证神经元细胞正常运转的后勤保障系统。
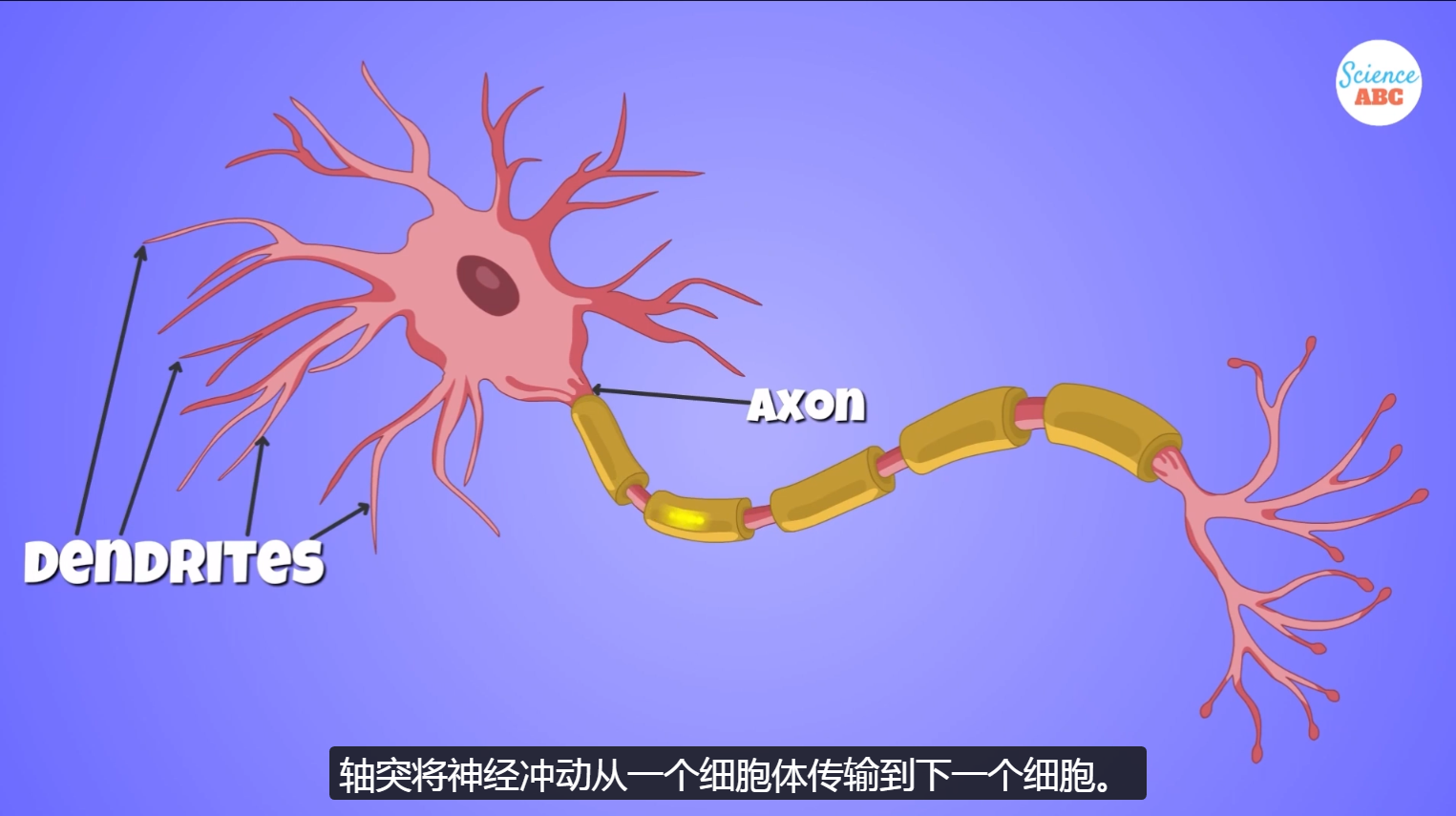
1、神经元的基本结构
神经元细胞主要由细胞体、轴突、树突三部分构成。
(1)细胞体
细胞体是神经元的代谢和营养中心,合成神经元所需的蛋白质和能量物质。
(2)轴突——发送器
顾名思义,一条比较长、分枝少的轴(为方便记忆,可以想象成一个高耸入云的电视信号塔,由于发送信号)。轴突的作用是发送神经递质。
轴突的起始部位(即细胞体与轴突的连接部位)称为轴丘。这个位置很重要,其钠离子通道密度较高,是静息电位 向 动作电位转换的 触发位点。
部分神经元轴突外会包裹一层髓鞘,能够加快神经冲动的传导速度。
(3)树突——接收器
细胞体向外延伸出的短、多分支的突起。 顾名思义,像一棵枝繁叶密的大树。树突的作用是接收临近神经元的轴突 发送过来的神经递质。
这三个概念请牢牢记住,下面需要反复提及。
2、突触
神经元之间并不直接相连,在突触中,通过电信号-化学物质-电信号 方式完成信号传递。
前文已叙,轴突是信号发送器,树突是信号接收器,具体如何发送接收呢?
轴突末梢的细胞膜中(称为突触前膜),内部含有储存神经递质的小泡。
树突或细胞体细胞膜上,分布着能与神经递质结合的受体(称为突触后膜)。
突触前膜释放神经递质,神经递质通过充满细胞液的突触间隙,与突触后膜结合。这种化学突触是神经元信息传递的主导方式。心肌细胞、平滑肌细胞、中枢神经系统中还存在电突触。
3、神经递质
目前被广泛公认的神经递质有50+种(按化学性质可清晰分类),主导神经系统绝大多数基础信号传递的核心递质约10种左右,承担了99%以上的神经传递,参与了从脊髓反射、骨骼肌收缩,到大脑的学习记忆、情绪调控、睡眠觉醒,再到自主神经系统功能调节的所有基础神经功能。分别为:
(1)兴奋性递质:谷氨酸(Glutamate)、乙酰胆碱(Acetylcholine / ACh)。
(2)抑制性递质:γ-氨基丁酸(GABA),甘氨酸(Glycine)。
(3)单胺:多巴胺(DA)、去甲肾上腺素(NE)、5-羟色胺(简写5-HT,又称血清素)、肾上腺素、组胺。
除此之外,还有:
(4)神经肽类:主要调节突触传递效率,而非直接传递信号。常见的为内啡肽和脑啡肽(镇痛物质,与阿片受体结合,缓解疼痛、产生愉悦感,内啡肽属于广谱强效,脑啡肽属于精准短效)、神经肽 Y(参与食欲调控、应激反应)、P 物质(参与痛觉传递、炎症反应)。
(5)嘌呤类神经递质:腺苷(Adenosine)(中枢抑制性递质,咖啡因、茶碱等物质的提神作用,就是通过阻断腺苷受体实现),三磷酸腺苷(ATP)(作为递质时,可直接作用于嘌呤能受体,介导快速的兴奋性突触传递;参与痛觉感知、突触可塑性调控,以及外周器官的功能调节;被释放后,可快速分解为腺苷)。
(6)气体类神经递质:一氧化氮,一氧化碳。
等等。
一些神经递质与神经元树突结合后,可以直接存储,比如去甲肾上腺素、多巴胺,直接被囊泡膜上的转运体摄取进入囊泡,重新储存,等待下一次被释放;一些神经递质无法存储,比如乙酰胆碱,被摄取后,经历酶促反应的转换,才能重新成为有活性的神经递质。
4、神经元电位
细胞膜内包含K⁺(钾离子)、Na⁺(钠离子)等正电荷,以及蛋白质阴离子(比如酶、结构蛋白等),电荷配平。
神经元的细胞膜含蛋白质形成的孔(称为离子通道),选择性允许某些离子(如钾离子、钠离子)通过。细胞内钾离子的浓度约为140-150mmol/L左右,细胞膜外钾离子浓度约为4-5mmol/L。由于浓度差异,钾离子 通过 钾漏通道 由细胞膜内部扩散至细胞膜外部(阴离子无法自由穿过细胞膜),导致膜内呈现负电。当电荷力和扩散力平衡时,进入稳定状态。理论上,若只存在钾离子,则对应的电位低于 -93mV(超级化电位)。
细胞内还含有钠离子。钠离子和钾离子恰恰相反,神经元胞浆内的钠离子浓度约为10-15mmol/L,细胞膜外钠离子浓度约为140-150mmol/L。钠离子由膜外向膜内扩散(但静息状态下,细胞膜对钾离子的通透性远高于钠离子)。钠离子的向内扩散抵消了一定钾离子造成的电位差;神经元安静时的基础电位,为 -70mV(静息电位)。
谷氨酸等兴奋性递质与树突、胞体膜结合后,开放非特异性阳离子通道,轴丘处的电位一旦达到 -55mV(阈电位),开启去极化,电压门控钠离子通道大量开放,达到 +30mV(动作电位峰电位),随后钠离子通道失活,钾离子通道大量开放,钾离子外流,直接达到-80mV— -90mV的超级化电位。
随后,细胞膜回到静息状态的离子通透性,由于电荷力>扩散力,钾离子通过漏钾通道内流,逐渐回到静息电位。
以上就是 静息电位—>阈电位—>动作电位—>超极化—>静息电位的循环过程。
可能诸位有疑问,按照上面的过程,膜内钾离子、钠离子不都乱套了吗(细胞膜内钾离子减少,钠离子增多)?这里面有两个因素:一是外流的钾离子仅占细胞内总钾离子的不足0.1%,不会显著改变细胞内外的离子浓度梯度。二是钠钾泵持续工作,持续将 3个钠离子泵出细胞、2 个钾离子泵入细胞,逐步恢复膜内外的离子浓度梯度。
这里需要说明的是:静息电位是指神经元全细胞膜的基础电位,安静状态下,胞体、树突、轴突的膜上都存在 -70mV 左右的静息电位。动作电位触发在轴丘,扩布至胞体。
5、突触后电位
前文介绍的神经元电位,是神经元整合信号后产生的“全域电位反应”,是一个宽泛的概念。下文要讲的突触电位,是神经元接收的输入信号时产生的局部电位。
依据功能突触后电位分为两种: 兴奋性突触后电位(EPSP)和抑制性突触后电位(IPSP)。
(1)兴奋性突触后电位(EPSP)
突触的过程为:
步骤一,上一个神经元释放神经递质:神经元的动作电位传导至轴突末梢,触发电压门控钙离子通道开放,细胞外的 钙离子流入末梢,内流的Ca²⁺会与突触前末梢内的钙结合蛋白(如突触结合蛋白)结合,触发突触小泡与突触前膜的融合,将小泡内的神经递质(如谷氨酸)释放到突触间隙。
步骤二,临近神经元接收神经递质:突出间隙的谷氨酸扩散至突触后膜(即下一个神经元的胞体膜、树突),与下一个神经元胞体膜、树突上的谷氨酸受体结合,受体构象发生改变,直接打开非选择性阳离子通道,导致Na⁺、Ca²⁺内流,以及部分K⁺外流,突触后膜电位从静息电位向极化方向偏移(电位从 -70mV 向 -55mV 靠近)。单个EPSP幅度很小,仅 1~5mV,此时的突触电位仅限于突触后膜局部位置,不会自动扩散到整个神经元,而是随距离衰减方式从树突 / 胞体向轴丘传递。
步骤三,触发动作电位:多个树突上的EPSP同时产生,它们的电位会叠加,向轴丘传递,轴突电位逐步升高,一旦轴丘电位突破 -55mV,就会触发动作电位,电压门控Na⁺通道大量开放,Na⁺快速内流,引发极化(即上文介绍的,神经元电位从静息电位到动作电位转换)。
(2)抑制性突触后电位 IPSP
和兴奋性突触后电位(EPSP)的过程基本相反。
抑制性神经递质(如GABA)被释放至突触间隙,结合下一个神经元树突/胞体的受体后,受体构象改变,Cl⁻通道开放;在生理状态下,细胞外Cl⁻浓度高于细胞内,浓度差驱动Cl⁻从细胞外流入细胞内,导致突触后膜超极化(电位从 -70mV 降到 -80mV 左右)。
(3)突触后电位
神经元同时接收来自多个不同突触的信号。比如,多个树突上的EPSP同时产生,它们的电位会叠加,让轴丘的去极化幅度快速增大;若此时有IPSP ,会抵消EPSP 的去极化。
这些EPSP、IPSP整合叠加后:①如果导致神经元轴丘电位≥阈电位,触发动作电位, 膜电位从 -55mV 飙升至 +30mV,随后经极化、短暂超极化,恢复到静息电位。动作电位会沿轴突长距离传导,同时反向扩布到胞体,引发整个神经元的电位剧变。② 整合叠加后轴丘电位<阈电位,无法触发动作电位,叠加的突触电位会逐渐衰减消失,神经元电位回到静息电位。③若 IPSP占主导,神经元会维持超极化状态,兴奋性降低。
6、知识串联
把上面只是串联一下,神经元信号连接、传递原理:
神经元静息状态,维持 -70mV的电位 —> 树突、胞体接收周边神经元释放的神经介质,由于Na⁺、Ca²⁺内流、K⁺外流、Cl⁻内流等因素,形成突触电位,传导至轴丘—> 当轴丘处叠加的电位 若高于阈电位,直接引发动作电位,打开电压门控钠离子通道,Na⁺内流,引发极化—>神经元电位直接变为 30mV —> 动作电位会以沿着轴突快速传导至轴突末梢,突触小泡的神经递质释放到突触间隙 —> 周围神经元的树突、胞体接收神经介质。
正常情况下,每次动作电位,都会引发神经递质释放。也有失效的情况:①如果轴突出现损伤、髓鞘脱失,动作电位可能在传导过程中衰减、阻滞,无法到达突触前末梢;②如果突触前末梢的电压门控Ca²⁺通道出现功能缺陷(如基因突变、药物阻断),递质释放的关键环节会断裂;③中枢神经系统中存在突触前抑制机制,比如一个抑制性神经元的突触会连接在兴奋性神经元的突触前末梢上,释放递质(如 GABA),导致突触前末梢发生轻度超极化,或减少电压门控Ca²⁺通道的开放数量。以上均会造成动作电位却无法释放神经递质的情况。
7、神经元细胞与神经递质
这里可能大家有一个疑问:动作电位—>释放神经递质,神经递质有50种以上,那么神经元细胞具体释放哪种神经递质呢?
根据戴尔原则,绝大多数神经元终生只释放一种主要的神经递质(如谷氨酸能神经元只释放谷氨酸)。 少数神经元会在释放主要递质的同时,共存释放神经肽(如脑啡肽、神经肽 Y)作为调质,调节突触传递的效率。
(1)谷氨酸(Glutamate)由绝大多数兴奋性神经元(如大脑皮层、海马的锥体神经元)的轴突末梢释放。
(2)乙酰胆碱(ACh):由基底前脑胆碱能神经元释放,参与学习记忆、觉醒等功能;此外,运动神经元、自主神经节释放乙酰胆碱,支配骨骼肌收缩,调控内脏活动。
(3)γ- 氨基丁酸(GABA):由抑制性中间神经元(如大脑皮层、小脑的篮状细胞)的轴突末梢释放。
(4)甘氨酸(Glycine):由脊髓和脑干的抑制性神经元中释放,是脊髓内的主要抑制性递质,参与躯体运动的调控。
(5)多巴胺(DA):由中脑多巴胺能神经元释放,参与运动控制、奖赏动机。
(6)血清素(5 - 羟色胺/5-HT):由脑干中缝核神经元释放,调节情绪、睡眠、食欲。
(7)去甲肾上腺素(NE):由脑干蓝斑核神经元释放,参与注意力、应激反应调节。
下一个问题,既然一种神经元,释放时只能释放一种神经递质,那接收时呢?能接受多种神经递质吗?答案是可以。比如脑皮层的锥体神经元(释放谷氨酸),其突触后膜上同时存在 谷氨酸受体、GABA 受体(抑制性信号),多巴胺受体、血清素受体等。
神经元基础知识讲解完毕。大家可以消化一下,把上面原理看明白了,理解脑机接口、神经疾病也就容易的多。
二、脑机接口(BCI)
脑机接口,即通过侵入、半嵌入、非侵入方式,采集脑电波信号,随后将采集到的信号进行解码,外部设备执行指令,从而实现意念操控。
截至目前,侵入式脑机接口,全球商业产品(如马斯克的Neuralink)约20例,科研临床试验约300-500例;非侵入式因无需手术,应用已达数十万例。 目前脑机接口成熟应用的在于医疗康复领域,比如高位截瘫患者用意念拧瓶盖、进食,四肢截肢者下棋、玩赛车游戏,渐冻症患者意念打字,脑卒中患者上肢功能恢复等。
1、脑电波(EEG信号)
脑机接口的本质是利用电极采集脑电波,脑电波如何来的?
认真看完前文的小伙伴估计会有猜测,脑波是由神经元静息电位—>动作电位转换引发的?这是一个常见的误解,是错误的。
目前医学界公认的脑波产生机制是突触后电位学说(否则前文为什么在写“4、神经元电位”后,专门又写“5、突触后电位”),而非动作电位学说。
回顾一下前文,突触后电位是如何形成的:神经元胞体膜和树突上的受体与神经递质结合后,受体构象发生改变,Na⁺、Ca²⁺内流、部分K⁺外流(受体结合兴奋性递质,从而引发的兴奋性突触后电位),或者Cl⁻内流(受体结合抑制性递质,从而引发的抑制性突触后电位),叠加后形成某个电位(当这种电位传导至轴丘,达到阈值,才会引发动作电位)。单个EPSP幅度、 IPSP很小,单个神经元含有多个树突形成的电位会进行叠加;当数百万甚至上亿个神经元同步活动时,它们的电信号会叠加形成宏观电场,这种电场通过脑组织、颅骨、头皮传导到体表,被电极捕捉后,就是我们说的 “脑电波”。
具体的,大脑皮层的锥体细胞排列整齐,其顶树突朝向皮层表面,当大量锥体细胞同步化地接受突触输入时,它们的突触后电位会进行叠加。这种同步化的群体电位变化强度足够大,能够穿透颅骨和头皮,被脑电图电极捕捉到。
2、脑电波分类
根据频率和振幅的不同,脑电波分为4种基础类型:
① <4Hz,称为δ波(睡眠、婴儿期、重度脑损伤);
② 4-7Hz,称为 θ波(浅睡眠、困倦、学习记忆)(海马主导);
③ 8-13Hz,称为 α波(清醒放松、闭眼状态);
④ 14-30Hz,称为 β波(清醒警觉、专注思考、运动控制)。
在这4中基础类型之外,还有一些补充的扩展波形,比如:
⑤ γ 波,频率可以达到30-100Hz,甚至更高,属于β波的高频延伸,是大脑高度专注、认知加工时的节律,比如人在解复杂数学题、识别面部时,前额叶和颞叶会出现 γ 波。
⑥ μ 波,频率8-13Hz,和α波一致,但仅在运动皮层区域出现。当人放松不动时μ波明显,一旦准备或执行肢体动作,μ 波会迅速抑制消失,常被用于运动意图的研究。
⑦病理波形,大脑异常放电,比如癫痫发作时,神经元会出现突发性、高振幅的同步放电,形成棘波(持续 20-70ms)或尖波(持续 70-200ms),这类波形是临床诊断癫痫的关键依据。
3、事件相关电位
之前提到的脑波,属于持续节律性脑电,是大脑在不同生理状态下的背景电活动,有固定的频率范围,持续存在。
大脑在特定事件/任务触发下,会产生的一过性电反应,没有固定频率,只在“准备动作、接收刺激”等特定场景下出现,叠加在基础脑波的背景上,称为事件相关电位(ERP)。
目前常见的被破译并应用于临床或脑机接口的时间相关电位包括:
(1)运动准备电位(MRCP):当受试者准备执行肢体动作/想象动作,动作前 1-2 秒基础脑波中会叠加出现缓慢负向电位,幅值与运动意图强度正相关。脑瘫患者意念控制机械臂,即利用运动准备电位。
(2)P300波: 当受试者识别到目标刺激(如看到自己需要的字),刺激出现后的300ms左右出现的基础脑波中会叠加出现正向电位。渐冻症患者沟通工具即利用P300波。
(3)稳态视觉诱发电位(SSVEP):当受试者注视特定频率闪烁的视觉刺激时,基础脑波会叠加产生与闪烁频率完全同步的电位。意念控制智能家居、脑机接口打字即利用稳态视觉诱发电位。
(4)错误相关负波(ERN): 当受试者意识到自己犯了错误,错误发生后50-100ms,基础脑波会叠加出现负向电位。 训练飞行员的错误规避能力即利用错误相关负波。
(5)体感诱发电位(SEP) :当受试者身体某部位受到痛觉刺激,刺激后几十到几百毫秒基础脑波会叠加出现系列电位。可用于诊断周围神经病变、判断神经传导是否正常。
(6)N400波:当受试者接收到语义异常信息、不符合常识的话,400ms左右后基础脑波会叠加出现负向电位,语义越异常幅值越大。可用于检测植物人/重度意识障碍患者的语言理解能力、研究儿童语言发育。
(7)视觉诱发电位(VEP):当受试者眼睛接收到特定视觉刺激(如闪光),刺激后100ms左右基础脑波会叠加出现正向电位(P100波),可用于诊断视神经病变、光眼早期筛查。
基础脑波、事件相关电位,是非侵入式脑机接口的核心信号来源。
4、侵入式脑机接口信号来源
相较而言,侵入式脑机接口信号来源更加丰富,能够探测到更微观的神经电信号,建立微观神经电信号与行为指令的关系。
侵入式BCI的核心信号是 单细胞动作电位(Spike)。这是Neuralink等侵入式BCI的核心依赖信号,也是目前解码精度最高的神经信号。
基础脑波是数千至数百万神经元的同步电位总和,是“宏观背景”;而Spike是单个神经元的精准放电信号,属于“微观指令”。
侵入式BCI可识别单个神经元兴奋时产生的电脉冲(持续时间短,可能只有1-2ms),且侵入式BCI空间分辨率更细微,可达10-20μm,能精准捕捉运动皮层中特定神经元的放电,从而解析出如“控制左手移动”、“手指抓握”时神经元的信号。 比如,Neuralink公司侵入式BCI,实现意念玩马里奥赛车游戏,就是对不同运动意图(如“左转”、“加速”)对应不同神经元集群的放电频率、时序组合,算法通过建立“放电模式→运动指令”的映射,实现更加细微的意念控制。
除了单细胞动作电位(Spike)外,还存在局部场电位(LFP),探测神经元集群在直径几百微米内产生的突触后电位总和,介于Spick和基础脑波之间,可作为辅助核心信号。
5、信号解析路还很漫长
目前,已明确破译的脑电信号,仅覆盖简单、低层级事件;部分破译的脑电信号,属于概率性关联,准确率只有50%-70%,无法支撑精准的意图控制,仅能用于辅助判断。
大脑活动的绝大部分脑电信号尚未破译。 比如复杂认知活动,思考数学公式、分析逻辑问题、制定计划等,这类活动涉及前额叶、顶叶、颞叶等多个脑区的协同工作,没有统一的信号特征,无法建立“信号模式→思维内容” 的映射。 再比如抽象思维与创意活动,产生一个创意点子、理解一首诗的意境,这类活动是大脑的高级功能,信号是多维度、动态变化的,现有技术无法捕捉其核心特征。 再比如复杂社交与情感活动,涉及边缘系统、前额叶等多个脑区的交互,信号极其复杂。
脑机接口的现有应用,多为基于已破译的简单信号,而复杂思维解码,还处于科学探索的早期阶段,短期内难以突破。
6、侵入式的问题
2024年5月,Neuralink首位人类受试者诺兰・阿博(颈部以下瘫痪)植入N1设备,后期85%电极丝回缩,不再与神经元有效接触。这里面原因较多,比如头部微小移动导致电极丝移位/脱落,柔性电极与脑组织长期摩擦;脑组织对异物的免疫反应,形成胶质瘢痕包裹电极;超细电极丝(4-6μm)长期使用后出现物理疲劳。
Neuralink随后对各方面进行了完善:电极设计改为石墨烯涂层和可降解材料,优化电极固定方式,延长电极寿命;手术技术采用穿刺,实现更精准植入;研发自修复材料和自适应电极阵列;改进系统算法。但电极长期稳定性仍待观察。
7、国内企业
我个人认为,脑机接口长期看,拥有巨大的发展潜力。但同时,目前国内上市公司没有太好的标的。
脑机接口头部研究机构,主要集中在高校(天津大学海河实验室、浙江大学、清华大学、南京大学、电子科技大学等)、中科院系统等科研机构。
医疗机构方面,30家左右医院可开展侵入式临床试验;$三博脑科(SZ301293)$ 是其中一家(但也仅仅是跟着喝口汤而已;而且只是因为其为上市公司而受关注,真正在医院领域里排不上);全国120多家医院可以开展非侵入式临床试验。
三、神经系统疾病
基于神经元的相关知识,会发现神经疾病多数和神经递质有关。
按照医保目录中,神经系统疾病用药的分类,主要包括精神安定药、精神兴奋药、抗帕金森氏病药物、抗癫痫药等几类。

1、抑郁症
此前写过一篇文章,抑郁症,丽珠加油。抑郁症的发病机理假说是单胺类神经递质失衡假说,认为大脑内5 - 羟色胺(5-HT)、去甲肾上腺素(NE)、多巴胺(DA) 三种单胺类递质的含量或功能下降,导致情绪调节能力受损。
目前主流药物为选择性单胺再摄取抑制剂。比如5-羟色胺再摄取抑制剂(SSRI)——氟西汀、帕罗西汀、舍曲林、氟伏沙明、西酞普兰等;5-羟色胺和去甲肾上腺素再摄取抑制剂(SNRI)——文拉法辛、度洛西汀、米那普仑等;多巴胺和去甲肾上腺素再摄取抑制剂(NDRI)安非他酮等。
这里的5-HT再摄取抑制是个什么概念呢?
神经元轴突释放5-HT到突触间隙后,一部分被临近神经元的树突/胞膜接收,但还有一部分并不会被接收,反而是被回收——释放5-HT的轴突触前膜上的 “5-HT 转运体(SERT)” 主动将5-HT转运回突触前神经元内,重新储存到突触小泡,等待下一次释放,实现循环利用(还有小部分未被回收的 5-HT,会被突触间隙的酶分解灭活)。这个回收,即再摄取。
再摄取抑制,就是阻断回收。具体机理为:5-HT转运体有两个关键结合点位:一个是5-HT 结合位点,专门识别并结合5-HT分子,另一个是钠离子结合位点。当突触间隙的5-HT 与 5-HT转运体的5-HT结合位点结合,且钠离子同时结合到对应位点时,5-HT转运体会发生构象变化,将5-HT分子从突触间隙拉回到突触前神经元内部。5-HT再摄取抑制剂的分子结构与5-HT具有相似性,比5-HT更高效地结合到 5-HT转运体的5-HT专属结合位点上,霸占了天然5-HT分子的位置,无法再发生转运所需的构象变化。这就导致5-HT会持续停留在突触间隙里(而不是被回收);相应的,突触后膜的5-HT受体能接触到更多、更持久的5-HT分子。
目前,抑郁症最新药物为2022年获批的盐酸托鲁地文拉法辛 和 右美沙芬安非他酮复方。盐酸托鲁地文拉法辛为5-HT、NE和DA再摄取抑制剂。
2、精神分裂症
精神分裂症的主流假说为多巴胺假说(多巴胺过量)和谷氨酸假说(谷氨酸受体、尤其是 NMDA 受体的功能低下,导致精神分裂症发病)。
突触前神经元兴奋,释放多巴胺到突触间隙(神经元之间的微小间隙);多巴胺与突触后膜的多巴胺受体(D1、D2、D3 等,其中 D2 是精神分裂症的核心靶点)结合,传递神经信号;未结合的多巴胺会被突触前膜的多巴胺转运体(DAT) 重摄取回神经元内,或被酶分解,从而终止信号,保证突触传递的平衡。
①精神分裂症阳性症状的特定脑区(中脑 - 边缘多巴胺通路),突触间隙的多巴胺无法被正常转回、清除,导致突触间隙多巴胺堆积;此外,突触后膜的D2 受体数量增多/敏感性增高,少量多巴胺也能触发过强的神经信号。②精神分裂症阴性症状的特定脑区(中脑 - 皮质多巴胺通路),此通路多巴胺能传递不足。
理解了这个原理,就方便理解治疗药物。以阿立哌唑为例:突触间隙的多巴胺 与 突触后膜的多巴胺D2受体结合点位结合后,会产生强信号;阿立哌唑可以和突触后膜的多巴胺D2受体结合,结合后触发弱信号。在中脑 - 边缘多巴胺通路,由于阿立哌唑抢占了一些受体结合点位,而与这些结合点位结合时发出的信号弱,所以整体上,起到了抑制作用。但在中脑 - 皮质多巴胺通路,突触间隙中的多巴胺量少,突触后膜大部分多巴胺D2受体望眼欲穿盼不来多巴胺,此时阿立哌唑与之结合,产生了弱信号,因此起到了一定的激动剂作用。
3、阿尔茨海默症
(1)乙酰胆碱酯酶抑制剂
2020年之前,FDA批准的阿尔茨海默症药物只有两类:乙酰胆碱酯酶抑制剂和美金刚。直到2020年之后,才陆续有阿杜那单抗(已撤回上市)、 仑卡奈单抗、多奈单抗获批,这三款单抗和神经递质无关,主要是为了清除β样淀粉形成的老年斑。在过往的药物中,主要针对神经递质乙酰胆碱。
乙酰胆碱(ACh)是神经中枢广泛存在的神经递质,基底前脑→大脑皮层 / 海马的通路的中枢胆碱能通路,和学习、记忆、认知功能密切相关。阿尔茨海默症患者的大脑中,基底前脑的胆碱能神经元会发生进行性退变、死亡,导致乙酰胆碱的合成、释放量大幅减少,突触间隙的乙酰胆碱浓度显著降低,无法有效与突触后膜的胆碱能受体结合。
还是回到此前的套路:突触前膜释放乙酰胆碱到突触间隙,突触后膜上的乙酰胆碱受体与乙酰胆碱结合。前面提到,乙酰胆碱不像5HT、多巴胺那样有转回机制,不会转回,而是被突触间隙的乙酰胆碱酯酶(AChE)快速水解降解为胆碱 + 乙酸。
药物乙酰胆碱酯酶抑制剂的作用是:乙酰胆碱酯酶上有一个能特异性结合乙酰胆碱的催化位点,乙酰胆碱酯酶抑制剂能够直接和这个位点结合,导致乙酰胆碱酯酶无法再与乙酰胆碱结合,从而无法再去水解降解乙酰胆碱。乙酰胆碱不再被快速降解,而是在突触间隙 堆积,浓度提升,等待与突触后膜受体结合。
(2)美金刚
除了多款乙酰胆碱酯酶抑制剂外,治疗阿尔茨海默症还有一款药物美金刚,是由于阿尔茨海默症患者存在谷氨酸能系统亢进情况。
如前文所属,谷氨酸是主要的兴奋性神经递质,被释放至突触间隙后,与突触后膜的谷氨酸受体(NMDA 受体)结合。
要理解美金刚的药理,需要回忆一下前面内容:“谷氨酸扩散至突触后膜(即下一个神经元的胞体膜、树突),与下一个神经元胞体膜、树突上的谷氨酸受体受体结合,受体构象发生改变,直接打开非选择性阳离子通道,导致Na⁺、Ca²⁺内流,以及部分K⁺外流,突触后膜电位从静息电位向极化方向偏移(电位从 -70mV 向 -55mV 靠近)。”
上面介绍的再摄取抑制剂、阿立哌唑等药物,普遍是与受体上结合点位结合;美金刚不是这种机理。美金刚不与谷氨酸竞争 受体的结合位点,而是结合在受体的离子通道内部,直接阻断Ca²⁺离子流(静息状态下,通道被镁离子挡着;神经元极化时镁离子解离,钙离子内流;此时美金刚再去堵上通道),挡住钙离子内流,自然把突触后膜电位变化降低,从而降低兴奋性突触后电位。
4、癫痫
癫痫的核心病理机制是大脑神经元异常同步化放电,通常是因为 兴奋性递质谷氨酸过度亢进、抑制性递质γ- 氨基丁酸(GABA)功能不足,二者的失衡是绝大多数癫痫发作的根本递质原因;此外,乙酰胆碱、多巴胺、5 - 羟色胺等递质会辅助调控这一平衡,在特定类型癫痫中发挥次要作用。
这么一看,美金刚不是谷氨酸受体拮抗剂吗?那是否可以治疗癫痫呢?不行。
突触后膜上,能与突触间隙的谷氨酸结合的受体有多个,主要为AMPA受体(占比70%以上)、NMDA受体、KA受体(三者均为离子型受体),以及代谢型谷氨酸受体(G 蛋白偶联受体(GPCR)的一种)。其中,KA受体的亲和力最强,AMPA受体亲和力中等,NMDA受体亲和力最低,且有镁离子门控的额外条件。
AMPA 受体仅介导Na⁺/K⁺跨膜流动,无 Ca²⁺内流,是癫痫异常放电的启动源。
既然癫痫的发病原因是兴奋性递质亢进、抑制性递质(γ- 氨基丁酸,即GABA)不足,那么治疗癫痫的药物也主要围绕此展开。
(1)增强γ- 氨基丁酸角度:
提升 GABA 浓度、增强 GABA 受体作用、让神经元超极化(膜电位更负),此类药物包括地西泮、氯硝西泮(直接结合 GABAA 受体,增强氯离子通道开放效率 / 时长,让更多 Cl⁻内流,强化神经元超极化)、氨己烯酸、托吡酯(抑制 GABA 降解酶,从而提升GABA浓度)、丙戊酸钠、加巴喷丁(促进 GABA 合成 / 释放)。
(2)阻断电压门控钠通道
既然AMPA 受体介导Na⁺/K⁺跨膜流动(无 Ca²⁺内流),是癫痫异常放电的启动源,那就阻断 Na⁺内流。
主要药物包括卡马西平、奥卡西平(选择性阻断神经元去极化时的钠通道开放,阻止 Na⁺内流)、拉莫三嗪(阻断钠通道 +轻度抑制谷氨酸释放)。
(3)抑制谷氨酸兴奋性通路
如吡仑帕奈,选择性阻断突触后膜 AMPA 受体。
(4)其他
例如左乙拉西坦,直接结合突触囊泡蛋白 SV2A,抑制突触前膜神经递质(谷氨酸、GABA)的异常释放,稳定信号传递。
丽珠集团的KCNQ 2/3,本质是结合神经元膜上的 KCNQ2/3 亚型,加大K⁺外流,从而降低膜内电位(超极化),使神经元需要远强于正常的兴奋性刺激,才能去极化到阈电位产生放电,最终实现神经元基础兴奋性的显著降低。
5、渐冻症(ALS)
渐冻症核心病理为神经元进行性变性、凋亡,导致肌肉失去神经支配而逐渐萎缩、无力,最终累及呼吸肌引发呼吸衰竭。渐冻症患者都会出现一种情况:突触间隙谷氨酸过度堆积,过度激活运动神经元的谷氨酸受体,导致神经元钙超载、变性死亡。
回到此前的知识:谷氨酸扩散至突触后膜,与下一个神经元胞体膜、树突上的谷氨酸受体受体结合(AMPA受体、NMDA受体、KA受体),直接打开非选择性阳离子通道,导致Na⁺、Ca²⁺内流,以及部分K⁺外流,因此突触后膜电位从静息电位向极化方向偏移。
而渐冻症患者,蓄积的谷氨酸过度激活运动神经元上的AMPA 受体(AMPA受体仅介导Na⁺/K⁺跨膜流动,无Ca²⁺内流),引发神经元去极化并解锁 NMDA 受体,随后NMDA受体过度激活,导致大量 Ca²⁺内流,激活胞内钙依赖性蛋白酶、核酸酶,引发神经元骨架破坏、DNA 损伤,最终凋亡。
看到这里,前面的药物,左乙拉西坦(直接结合突触囊泡蛋白 SV2A,抑制突触前膜神经递质(谷氨酸、GABA)的异常释放)、吡仑帕奈(选择性阻断突触后膜 AMPA 受体)、美金刚(结合在受体的离子通道内部,直接阻断Ca²⁺离子流)不应该都有点用吗?实际临床上,左乙拉西坦确实可以用于对症处理 ALS 合并症,另外两个不行。
吡仑帕奈在国外做过临床试验,结果以失败告终。美金刚不行的原因在于,AMPA 受体原本不会导致钙离子内流,但AMPA受体持续激活,受体亚基重构(CP-AMPAR),导致AMPA 受体可直接介导少量 Ca²⁺内流;此外,AMPA受体受强刺激后会通过其他路径导致钙超载;美金刚的亲和力弱,在谷氨酸过量情况下,根本竞争不过。
FDA首个批准的ALS药物为利鲁唑。
回到前面的知识:①突触前膜发生的:神经元的动作电位传导至轴突末梢,触发电压门控钙离子通道开放,细胞外的 钙离子流入末梢,内流的Ca²⁺会与突触前末梢内的钙结合蛋结合,触发突触小泡与突触前膜的融合,将小泡内的神经递质(如谷氨酸)释放到突触间隙。②突出后膜发生的:谷氨酸扩散至突触后膜,与下一个神经元胞体膜、树突上的谷氨酸受体受体结合(AMPA受体、NMDA受体、KA受体),直接打开非选择性阳离子通道,导致Na⁺、Ca²⁺内流,以及部分K⁺外流,因此突触后膜电位从静息电位向极化方向偏移。
利鲁唑属于多个靶点:①利鲁唑通过阻断突触前膜的电压门控钙通道(VGCC),减少 Ca²⁺内流至突触前神经元末梢,从而抑制谷氨酸囊泡的释放,从源头减少突触间隙的谷氨酸浓度;②利鲁唑能选择性阻断中枢运动神经元上的电压门控钠通道,稳定运动神经元的静息膜电位,减少异常去极化和放电;③轻度、非竞争性拮抗突触后膜的AMPA受体和NMDA 受体,减少钙离子内流。
讲到这里,可能大家又有一个疑问:突触前膜、突触后膜在激活时都存在钙离子内流情况,那神经元细胞内的钙离子不就无限升高了吗?正常情况下,存在Na⁺-Ca²⁺交换体(泵出1个钙离子、泵入3个钠离子)和Ca²⁺-ATP 酶(消耗ATP,泵出1个钙离子,泵入2个氢离子)。
ALS这个疾病目前只能延缓,从机理上并没有完全搞清楚。
在这里,羡慕妒忌下@Lifuluan ,十几年前的老同事;人家离职后直接自己开了CRO;这几年再次创业,他们公司的ALS治疗产品已经获批IND:从技术路径角度,是FIC;从疗效角度,全球范围内所有上市+在研产品中,BIC。这人与人的差距咋就越来越大了呢?
6、抗焦虑
接着,大家猜一下,焦虑,到底咋用药呢?增强兴奋性还是增强抑制性?
估计有焦虑症的小伙伴都猜对了。焦虑的本质,不是压抑,而是情绪中枢过度兴奋。所以,抗焦虑的药物,是增强 GABA 的抑制性作用,和癫痫药物有一定的重叠。
话说,小作文,我一天能写好几篇;而这种科普型文章写着太费时费力了,写的头晕脑胀。本篇主要内容阐述完毕,先写到这里,回头再补充。